GeneCopoeia’s GeneHero™ CRISPR-Cas9 products and services provide a complete, powerful solution to your genome editing needs. Products and services include:
- CRISPR Plasmids
Transfect cells with our CRISPR plasmids with Cas9 and sgRNA for human, mouse, and rat. Search our database of more than 45,000 human, mouse, and rat genes for genome editing using CRISPR.
Recombinant Streptococcus pyogenes Cas9 nuclease, purified from E. coli, is a great choice for genome editing applications in vitro or in vivo.
- CRISPR Lentivirus
Genome integration of CRISPR elements using lentivirus. Cas9 and/or sgRNA packed in purified lentiviral particles at 108TU/ml, ready to infect all cell types.
- CRISPR AAV
Episomal expression of CRISPR components with adeno-associated viral particles carrying Cas9 and/or sgRNA, excellent for tissue and animal transduction.
Premade Cas9-expressing stable cell lines are great for sgRNA library screening and other high-throughput CRISPR-Cas9 applications.
Advantages
- RNA-guided genomic DNA recognition regardless of the methylation status
- Similar or greater gene-editing efficiency compared to ZFNs and TALENs
- Capable of editing multiple genes simultaneously (multiplexing)
- Simple and fast design process. No need to reengineer the nuclease for each new target
Product Information
CRISPR Plasmids
sgRNA expression plasmids are available for targeting virtually any gene in any experimental system. sgRNA plasmids express either sgRNA only or sgRNA + Cas9 nuclease in an all-in-one format. Lentiviral clones express sgRNA alone. Cas9-expressing lentiviral clones are available separately.
The CRISPR plasmid options listed below are intended for using 20 ntsgRNAs with the wild-type Cas9 nuclease from Streptococcus pyogenes(SpCas9). GeneCopoeia also provides alternate CRISPR options, such as Cas9 D10A nickase and other high-fidelity versions of Cas9, SaCas9, and shorter sgRNAs. To purchase reagents for these other options, please contact us for a custom quote.
Custom sgRNA Expression Plasmids
| Vector | Promoter | sgRNA and Cas9 | Selection Marker/ Reporter Gene | Vector Type | Vector Map |
|---|---|---|---|---|---|
| All-in-one (Cas9 nuclease + sgRNA) clones | |||||
| pCRISPR-CG02 | U6 | sgRNA and CBh-driven Cas9 (wild type) in the same vector | – / – | Non-viral | |
| pCRISPR-CG04 | U6 | sgRNA and CMV-driven Cas9 (wild type) in the same vector | Neomycin / copGFP | Non-viral | |
| pCRISPR-CG07 | U6 | sgRNA and CBh-driven Cas9 (wild type) in the same vector | – / copGFP | Non-viral | |
| pCRISPR-CG08 | U6 | sgRNA and CBh-driven Cas9 (wild type) in the same vector | – / mCherry | Non-viral | |
| pCRISPR-CG12 | U6 | sgRNA and CMV-driven Cas9 (wild type) in the same vector | Neomycin / mCherry | Non-viral | |
| pCRISPR-AD01 | U6 | sgRNA and CMV-driven SaCas9 in the same vector | – / | AAV | |
| pCRISPR-AD02 | U6 | sgRNA and hSyn-driven SaCas9 in the same vector | – / – | AAV | |
| pCRISPR-LvSG06 | U6 | sgRNA and EF1a-driven Cas9 (wild type) in the same vector | Puromycin / – | Lentiviral | |
| pCRISPR-LvSG09 | U6 | sgRNA and EF1a-driven Cas9 (wild type) in the same vector | Puromycin / eGFP | Lentiviral | |
| pCRISPR-LvSG10 | U6 | sgRNA and EF1a-driven Cas9 (wild type) in the same vector | Hygromycin / – | Lentiviral | |
| sgRNA only clones | |||||
| pCRISPR-SG01 | U6 | sgRNA only | Hygromycin / – | Non-viral | |
| pCRISPR-AV20 | U6 | sgRNA only | – / – | AAV | |
| pCRISPR-AV22 | U6 | sgRNA only | – / eGFP | AAV | |
| pCRISPR-LvSG03 | U6 | sgRNA only | Puromycin / mCherry | Lentiviral | |
Click here to search our database of sgRNA plasmids for knocking out more than 45,000 human, mouse, and rat genes.
Synthetic sgRNAs
GeneCopoeia offers customers a design and chemical synthesis service for sgRNAs (single-guide RNAs) targeting genes of interest. Chemically synthesised sgRNAs (including crRNA and tracrRNA) are purified via HPLC and modified by 2′-O-methyl and thiophosphate at the 3′ and 5′ ends. When Cas9 nuclease is present, the sgRNA recognises the target DNA sequence and guides Cas9 to cleave the target site. This creates double-strand breaks (DSBs) in the DNA and enables genome editing, including gene knockout, knock-in and mutation.Performance
| (A) | 
|
| (B) | 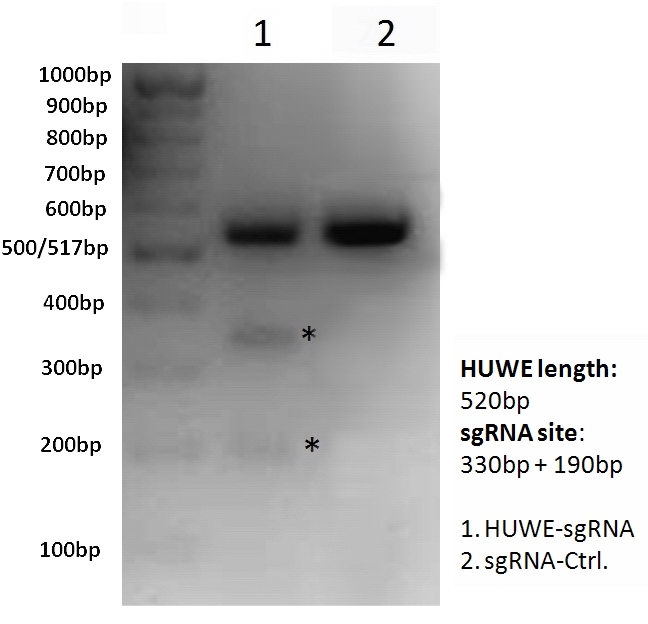 |
Fig.1 HUWE1-sgRNA/Cas9 targets the HUWE1 gene in HEK293T cells
(A) Design of HUWE1-sgRNA and genomic PCR primers; (B) Transfection of HEK293T cells in a 6-well plate using HUWE1-sgRNA/Cas9 clones and sgRNA/Cas9 control clones (lane 2). Cells were harvested 40 hours after transfection, genomic DNA was extracted, and PCR amplification was performed using HUWE1-specific PCR primers. The PCR products were gel-purified, mixed with buffer, denatured, annealed, and then digested with T7 ENI for 60 minutes in a 37℃ water bath. The fragment length of the HUWE1 PCR product should be 520 bp, and the products after T7 ENI digestion should be 330 bp and 190 bp, respectively.
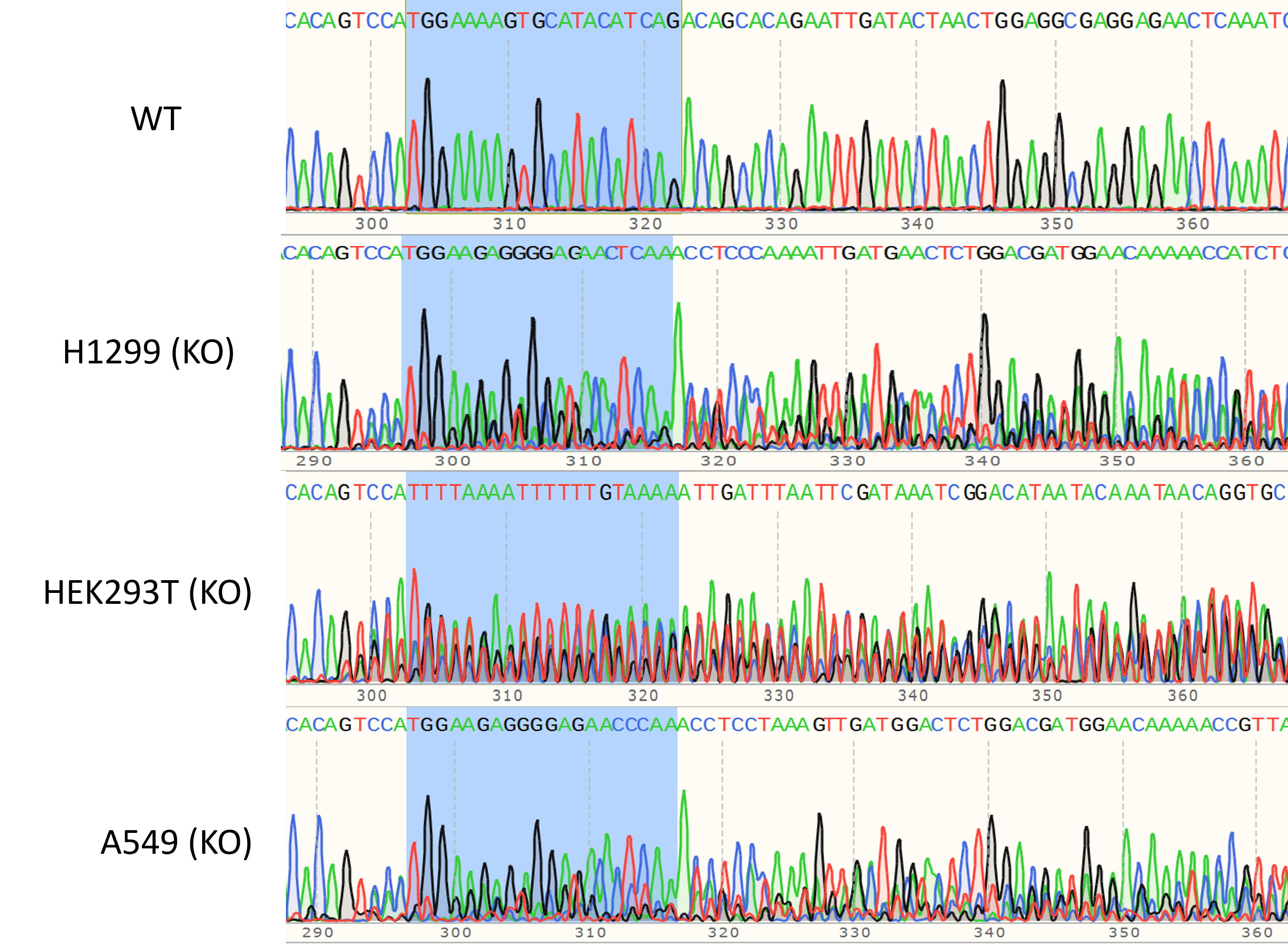
Fig.2 Chemically synthesized sgRNAs were used to generate FBXW7 knockout cell lines.
In a 24-well plate, Cas9-sgRNA complexes were transfected using CRISPR-Fectin™ transfection reagent (Cat# EF015), and cellular gDNA was sequenced 48 hours later.
CRISPR Lentivirus
Custom sgRNA Lentiviral Particles
sgRNA for human, mouse, and rat packaged in high-titer lentivirus.
| Vector | Promoter | sgRNA and Cas9 | Selection Marker/ Reporter Gene | Vector Type | Vector Map |
|---|---|---|---|---|---|
| pCRISPR-LvSG03 | U6 | sgRNA only | Puromycin / mCherry | Lentiviral | |
| pCRISPR-LvSG06 | U6 | sgRNA and EF1a-driven Cas9 (wild type) in the same vector | Puromycin | Lentiviral | |
| pCRISPR-LvSG09 | U6 | sgRNA and EF1a-driven Cas9 (wild type) in the same vector | Puromycin / eGFP | Lentiviral | |
| pCRISPR-LvSG10 | U6 | sgRNA and EF1a-driven Cas9 (wild type) in the same vector | Hygromycin | Lentiviral |
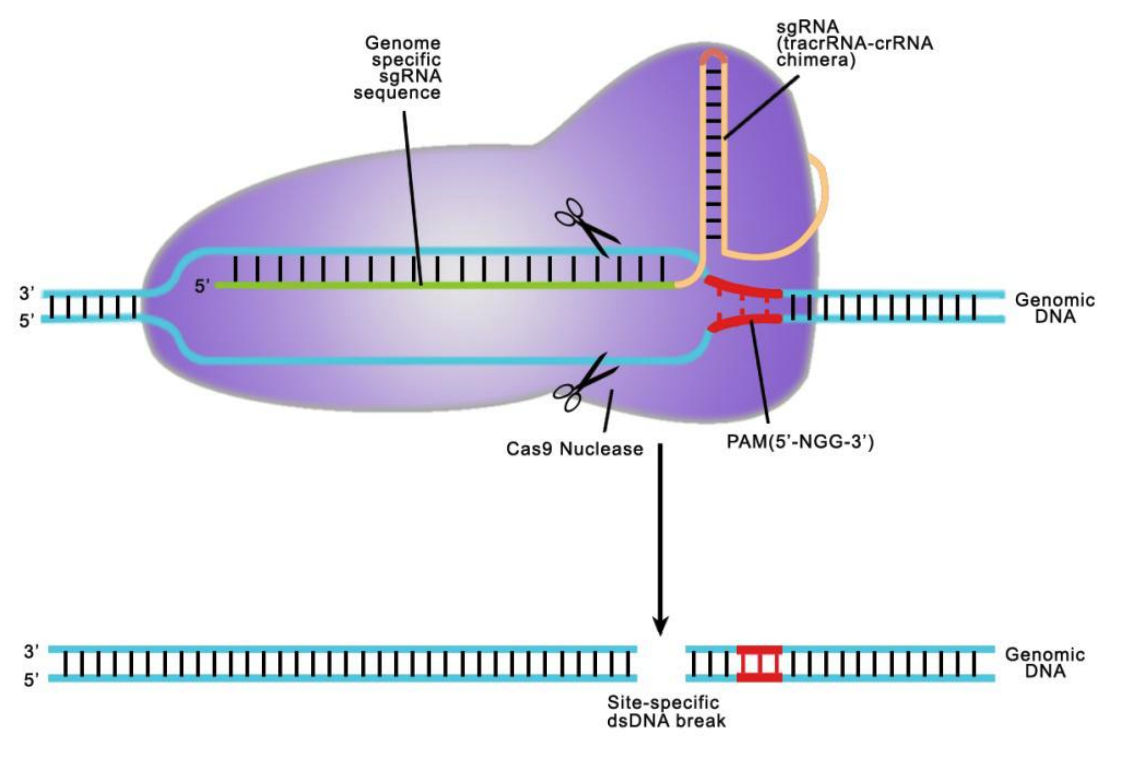
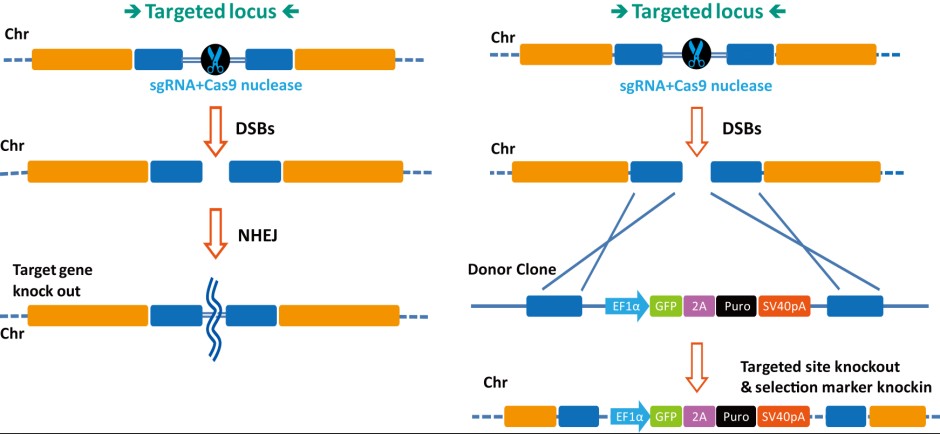
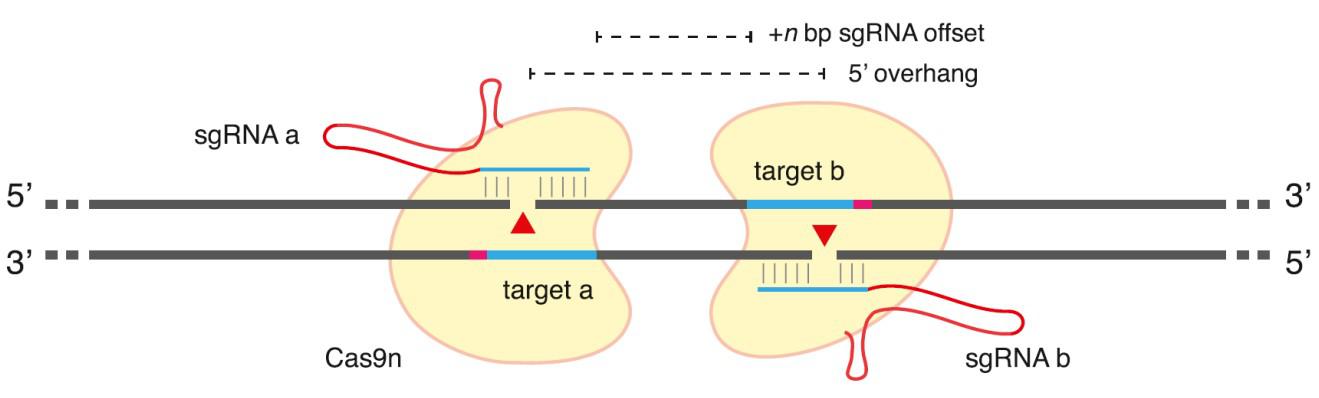 Figure 3. General scheme of Cas9 double-nickase strategy. From Ran, et al. (2013). Two additional strategies, the use of truncated (17-18 nucleotide) sgRNAs, as well as a Cas9-FokI fusion, also dramatically reduce CRISPR-mediated off-target genome modification. However, these methods suffer from even further reductions in on-target activity and/or more severe design constraints compared with the double nickase approach.
Figure 3. General scheme of Cas9 double-nickase strategy. From Ran, et al. (2013). Two additional strategies, the use of truncated (17-18 nucleotide) sgRNAs, as well as a Cas9-FokI fusion, also dramatically reduce CRISPR-mediated off-target genome modification. However, these methods suffer from even further reductions in on-target activity and/or more severe design constraints compared with the double nickase approach.