 |
Ed Davis, Ph.D. |
Introduction
A typical mammalian gene usually does not encode a single protein, thanks partly to the phenomenon of alternative mRNA splicing. Nearly all mammalian genes contain multiple introns, and greater than 90% of all intron-containing genes undergo alternative splicing to generate multiple transcript variants, and, subsequently, different protein isoforms (Pan, et al., 2008; Park, et al., 2018). Differential alternative splicing usually occurs within and between tissues, but some (5%) can occur between individuals (Kwan, et al., 2007; Wang, et al., 2008). As a result, one gene can potentially express many different proteins.
At GeneCopoeia, we provide customers with DNA-based tools that are used for many different types of gene function studies. These include plasmids for open reading frame (ORF) expression, gene knockout via CRISPR sgRNA, microRNA (miRNA) validation studies using 3 ’UTRs, etc.as well as qPCR primers. When customers request these reagents, they often encounter multiple accession numbers for each gene and do not know which one they need to order. Likewise, a customer might be interested in using plasmids to study a particular gene, but, when asked, they will not know the accession number of the variant or isoform they are working with. In this Technical Note, we talk about the multi-variant diversity of mammalian genes, and how the accession number of the gene you are working with needs to be a major consideration when requesting different types of plasmids from GeneCopoeia.
Considerations for different accession numbers in gene-based applications
As mentioned, most mammalian genes have multiple exons and are alternatively spliced, leading to the production of multiple proteins expressed from a single gene, as shown in Figure 1. One unusual exception to alternatively spliced genes is the human SOX2 gene. As of this writing in December 2019, the National Center for Biotechnology Information (NCBI) indicates that human SOX2 encodes only one known transcript, and this transcript contains only a single exon (https://www.ncbi.nlm.nih.gov/nuccore/NC_000003.12?report=genbank&from=181711925&to=181714436). Therefore, researchers do not need to know SOX2’s accession number (which happens to be NM_003106.4) when requesting plasmids or qPCR primers intended for studying this gene from GeneCopoeia. Easy, right?
Again, human SOX2 is an exception. Below, we discuss different types of gene-based applications and how they are affected by the presence of multiple transcript variants.
Accession number considerations for protein expression
One of the most widely used approaches for studying gene function is to express a protein from an ORF on a plasmid. This plasmid can be used to transfect cultured cells directly or be incorporated into a virus that can infect cells. It is important to keep in mind that alternative mRNA variation can affect a protein expressed from a plasmid in two different ways: 1) The length of the protein being produced; and 2) The availability of epitopes recognized by antibodies.
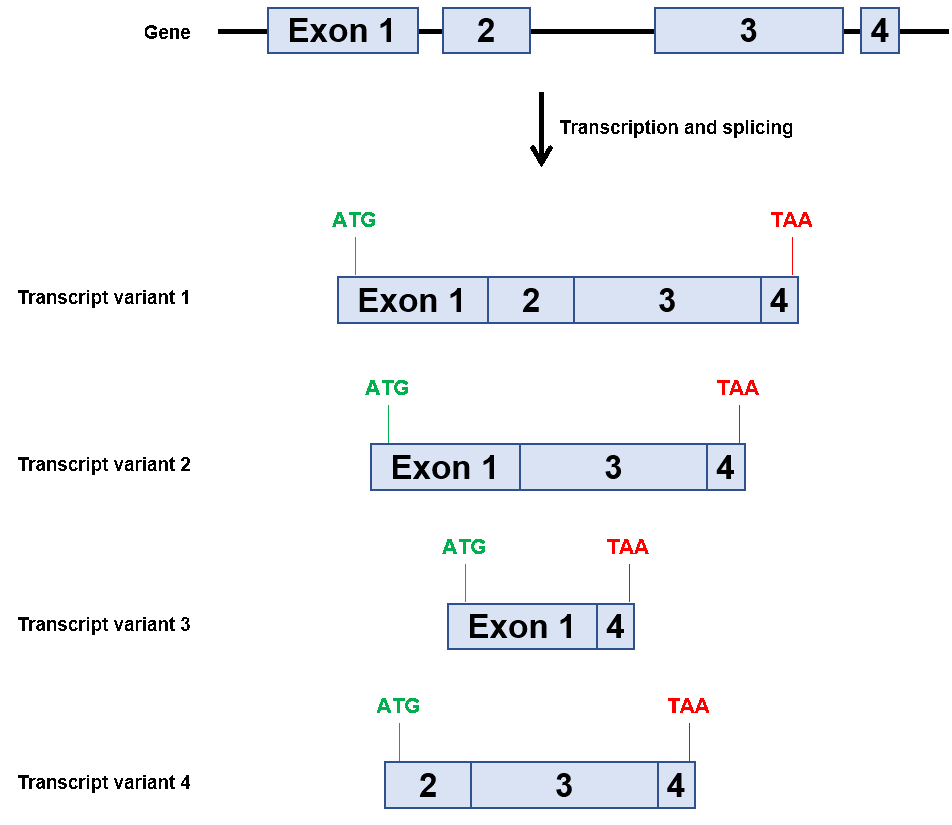
Figure 1. Example of alternative splicing of a hypothetical multi-exon gene.
One example of a gene that is highly affected by alternative splicing is human CD36. As of this writing in December 2019, CD36 has 16 known transcript variants (https://www.ncbi.nlm.nih.gov/nuccore/NC_000007.14?report=genbank&from=80602188&to=80679277). Some of these variants differ from one another only in the length of either their 5’ or 3’ UTRs. However, this gene undergoes alternative splicing to produce six distinct known proteins of lengths 472, 433, 412, 396, 438, and 317 amino acids. Four of these isoforms all have the same initiator methionine, whereas isoforms 4 and 6 have two different initiator methionines. All six proteins have the same C-terminal amino acid.
Accession number considerations for CRISPR-mediated knockout
One of the bedrock applications for CRISPR is gene knockout, whereby Cas9-mediated double strand breaks (DSBs) are repaired in error-prone fashion to produce frameshift mutations in protein-coding regions. If you want to use CRISPR to knock out a gene that encodes multiple protein isoforms, then you need to take the following considerations into account:
- Whether you want all possible protein isoforms knocked out
- Whether you want only one protein isoform knocked out.
If you need a knockout of all known protein isoforms of your gene, it might be possible to design one sgRNA to simultaneously knock out all isoforms. Such an sgRNA must be designed so that it targets a region of the mRNA in common with all known transcript variants. One way in which CRISPR sgRNAs are designed is to target as close to the initiator ATG of the gene as possible, because a frameshift mutation at that position of the gene is likely to cause the occurrence of a stop codon early in the gene sequence and knock it out. Alternatively, sgRNAs are also designed to target sequences encoding conserved and/or known essential functional domains. If your gene encodes multiple splice variants, and all splice variants have the same initiator ATG or critical domain, then it will be no problem to design a single knockout sgRNA using this strategy.
But what about the case of the CD36 gene? As mentioned, CD36 encodes transcript variants with three possible initiator ATGs. Protein isoforms 1, 2, 3, and 5 each are encoded by transcripts that have exactly the same first protein-coding exon. This exon is excluded from isoforms 4 and 6. In addition, the first protein-coding exon of isoform 4, which overlaps with the protein-coding regions of isoforms 1, 2, 3, and 5, is excluded from isoform 6 However, the second protein coding exon of isoform 6 is also found in each of the other five isoforms. Therefore, one sgRNA targeting the second protein-coding exon of the transcript encoding isoform 6 should be sufficient to knock out all known protein isoforms of CD36.
In some cases, though, you might be unable to find a region in common among all transcript variants. If so, then it will be necessary to use more than one sgRNA sequence to knock all of them out simultaneously.
Conversely, you might only want to knock out one of the isoforms and leave the others intact. Doing so requires that the isoform you are interested in contains a region that is unique to that isoform. In the case of CD36, none of the six known isoforms contain protein-coding sequence that is unique, so it would not be possible to knock out any one specific protein isoform by itself, if more than one isoform is expressed in the cells you are working with.
Accession number considerations for shRNA-mediated knockdown
shRNA-mediated gene knockdown differs from CRISPR in two fundamental ways: 1) CRISPR causes a permanent change to the genetic code in the chromosome, whereas shRNA does not, since the former acts on DNA while the latter acts on RNA; and 2) CRISPR is capable of causing a 100% elimination of the gene product, but shRNA usually does not. However, despite these differences, the considerations for transcript variants is the same. In some cases, a single shRNA will be able to knock down all transcript variants of a gene at once, while in other instances multiple shRNAs used together will be required. Likewise, it will only be possible to selectively knock down one transcript variant while leaving the other variants intact if that variant contains unique sequence.
Accession number considerations for fusion tagging
Another common gene function study application is fusion tagging, which typically involves adding another protein sequence, such as green fluorescent protein (GFP), or a small polypeptide like FLAG, to either the N- or C-terminus of the protein of interest.
Fusion tagging is usually accomplished in one of two ways. The first approach is to use an ORF-expressing plasmid, as mentioned earlier. If the gene expresses multiple protein isoforms, then you will need to know which isoform you are working with, so that you can choose the correct ORF plasmid.
The second approach to fusion tagging is to use CRISPR gene editing to place the tag on the gene at its native chromosomal locus. This approach can be advantageous over the ORF plasmid approach because it allows the gene to be expressed under its natural gene regulatory elements. However, due to the presence of alternative mRNA splicing, if you want to use CRISPR to place a fusion tag on your protein, you will again need to be aware of the existence of multiple transcript splice variants and protein isoforms for that gene, because in some cases, different isoforms for the same protein can have different N- or C-termini. One good example of such a gene is human ASXL1. As of this writing in December 2019, this gene encodes three known transcript variants, and respective protein isoforms, of 1,541, 85, and 1,480 amino acids (https://www.ncbi.nlm.nih.gov/nuccore/NC_000020.11?report=genbank&from=32358062&to=32439319). Among these three variants, there exist two different stop codons. If a researcher is interested in placing a C-terminal fusion tag on ASXL1, they would need to either decide to simultaneously tag isoforms 1 and 3 (which share the same C-terminal amino acid), or tag isoform 2. If they wanted to use CRISPR to tag all three isoforms of ASXL1 at the C-terminus, then they would need to use two sets of tagging reagents. In addition, there are two different initiator ATGs among the three different variants. One of these is shared between isoforms 1 and 2. So, a single N-terminal tag will not simultaneously tag all three isoforms either. Note, however, that tagging the N-terminus is not always a good idea. If the protein has a signal peptide, an N-terminal tag placed upstream of the signal peptide might get cleaved during post-translational processing, and so an N-terminal tag might need to be placed between the signal peptide and the rest of the protein
Accession number considerations for promoter studies
The next type of accession number situation to consider involves gene promoter studies. GeneCopoeia provides plasmids that express promoters to control the expression of a downstream reporter gene, such as Gaussia luciferase or GFP. Some genes with multiple transcript variants are expressed from a single promoter. Others, though, can have many possible promoters. One example of this is the human Smarca2 gene. As of this writing in December 2019, this gene has seven known transcript variants (https://www.ncbi.nlm.nih.gov/nuccore/NC_000009.12?report=genbank&from=2015347&to=2193624) and five different predicted promoters (https://www.genecopoeia.com/product/search3/?s=SMARCA2&search_type=1).
Accession number considerations for 3’ UTR studies
The 3’ untranslated region (3’ UTR) of a gene contains important regulatory elements, including the polyadenylation signal and binding sites for micro RNAs (miRNAs). GeneCopoeia also provides plasmids with 3’ UTRs placed downstream of a reporter gene, such as Gaussia luciferase. These 3’ UTR reporter plasmids are also affected by the existence of multiple transcript variants, since different transcripts from the same gene can have different 3’ UTRs. The human ASXL1 gene is also an excellent example of this. As mentioned above, this gene has three different known transcript variants, and among these transcripts there exist at least two distinct 3’ UTRs.
Accession number considerations for qPCR primers
The last example of a gene function study tool affected by the presence of multiple transcript variants is a qPCR primer pair, which is used to measure the relative abundance of mRNA produced by a single gene in cells. GeneCopoeia’s gene qPCR primers are typically designed to recognize exon-exon junctions that exist only in mature, spliced mRNA. Therefore, as with other applications, qPCR primers can potentially be designed to amplify all transcript variants, or only one specific transcript variant.
For the CD36 gene mentioned earlier, it is possible to design one or more qPCR primer pairs that will amplify the transcript variants encoding all known all protein isoforms simultaneously. Conversely, one primer pair can be designed to only amplify the transcript variants encoding four of the isoforms, but not the other two, and one primer pair can be designed that amplifies only the transcript variant encoding isoform 2 alone.
Conclusion
The phenotypic characteristics of organisms are influenced not only by the identity of the genes in its genome, but also by the diversity caused by alternative splicing variation occurring within genetic loci. While this diversity has obvious positive evolutionary benefits, it is also a potential source of confusion and error in gene-based research. Therefore, it is really important to know that a gene you are studying might express multiple protein isoforms, and that you should know the identity of the isoform you are working with. At GeneCopoeia, our scientists have a wealth of expertise with functional genomics applications in mammalian systems, and can help guide you to the gene-based product that suits your research requirements. We provide sequence-verified plasmid DNA, purified lentivirus, or purified adenoassociated virus, and we even offer complete, start-to-finish custom cell line construction services.
Want to know more about GeneCopoeia’s gene-based products and services or to place an order? Visit our website, call 1-301-762-0888, or email inquiry@genecopoeia.com.
References
- Kwan, et al. (2007). Heritability of alternative splicing in the human genome. Genome Research 17, 1210.
- Pan, et al., 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics 40, 1413.
- Park, et al. (2018). The Expanding Landscape of Alternative Splicing Variation in Human Populations. The American Journal of Human Genetics 102, 11.
- Wang, et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470.
| Copyright ©2019 GeneCopoeia, Inc. Email: inquiry@genecopoeia.com Tel: +1 (866) 360-9531 TNGE10-120319 |