Advantages
- All sgRNA-carrying lentiviral particles functionally validated for target cleavage of PD-1 or PD-L1
- 13 and 7 sgRNAs targeting all exons in PD-1 and PD-L1 respectively
- Next-day shipping, ready-to-use for the knockout of PD-1 or PD-L1 by CRISPR-Cas9 genome editing
- High titer (>108 TU/ml) lentiviral particles with mCherry reporter for in vitro and in vivo studies
Product Details


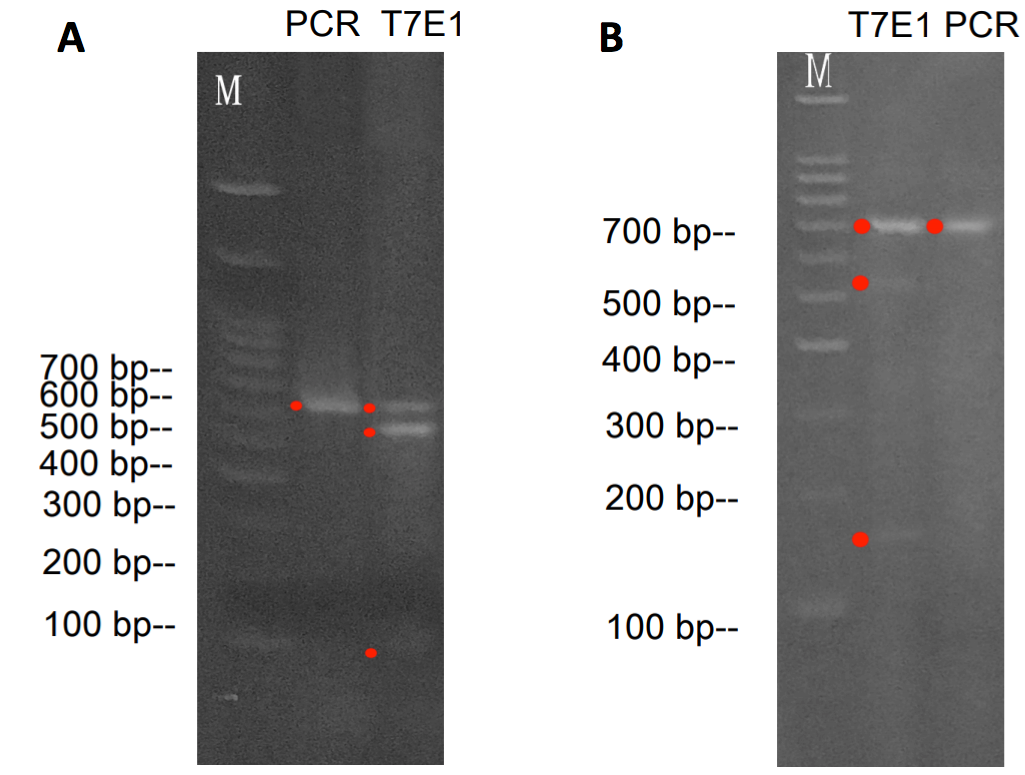 Figure 3. Validation of target cleavage by T7 endonuclease I (T7EI) assay. A. T7EI assay with the PD-1. A 631 bp PD-1 gene fragment from PCR was then tested by T7E1 Assay. The T7E1 cleavage resulted in two additional bands: one 515 bp and the other 116 bp. B. T7EI assay with the PD-L1. A 701 bp PD-L1 gene fragment from PCR was then tested by T7EI Assay. The T7EI cleavage resulted in two additional bands: one 535 bp and the other 166 bp.
Figure 3. Validation of target cleavage by T7 endonuclease I (T7EI) assay. A. T7EI assay with the PD-1. A 631 bp PD-1 gene fragment from PCR was then tested by T7E1 Assay. The T7E1 cleavage resulted in two additional bands: one 515 bp and the other 116 bp. B. T7EI assay with the PD-L1. A 701 bp PD-L1 gene fragment from PCR was then tested by T7EI Assay. The T7EI cleavage resulted in two additional bands: one 535 bp and the other 166 bp.
Ready-to-use purified lentiviral particles, used together with Cas9 nuclease clones, Cas9 lentiviral particles, Cas9 stable cell lines or premade labeled cancer cell lines for effective human PD-1, PD-L1 gene knockout in a variety of cell types including difficult-to-transfect cells, primary cells, stem cells and non-dividing cells as well as in vivo use for transgenic animals.
The sgRNA targeting PD-1 or PD-L1 is driven by U6 promoter. There is a mCherry reporter gene and a puromycin selection marker in the lentiviral genome, as illustrated in Figure 1. Transduction of the lentiviral particle can be visualized by mCherry protein using fluorescence microscopy as shown in Figure 2. Purified lentiviral particles are offered in above 3×108 TU/ml.
Figure 1. sgRNA carrying lentiviral genome.
Figure 2. Fluorescence microscopy images of HEK293 cells tranduced with lentiviral particles carrying sgRNA and mCherry reporter gene. A. HEK293 cells tranduced with lentiviral particles carrying sgRNA targeting PD-1 in bright field and RFP channel. B. HEK293 cells tranduced with lentiviral particles carrying sgRNA targeting PD-L1 in bright field and RFP channel.
Target recognition and cleavage of PD-1 and PD-L1 was verified using IndelCheckTM T7 endonuclease I assay as shown in Figure 3.
Figure 3. Validation of target cleavage by T7 endonuclease I (T7EI) assay. A. T7EI assay with the PD-1. A 631 bp PD-1 gene fragment from PCR was then tested by T7E1 Assay. The T7E1 cleavage resulted in two additional bands: one 515 bp and the other 116 bp. B. T7EI assay with the PD-L1. A 701 bp PD-L1 gene fragment from PCR was then tested by T7EI Assay. The T7EI cleavage resulted in two additional bands: one 535 bp and the other 166 bp.
Technology Overview
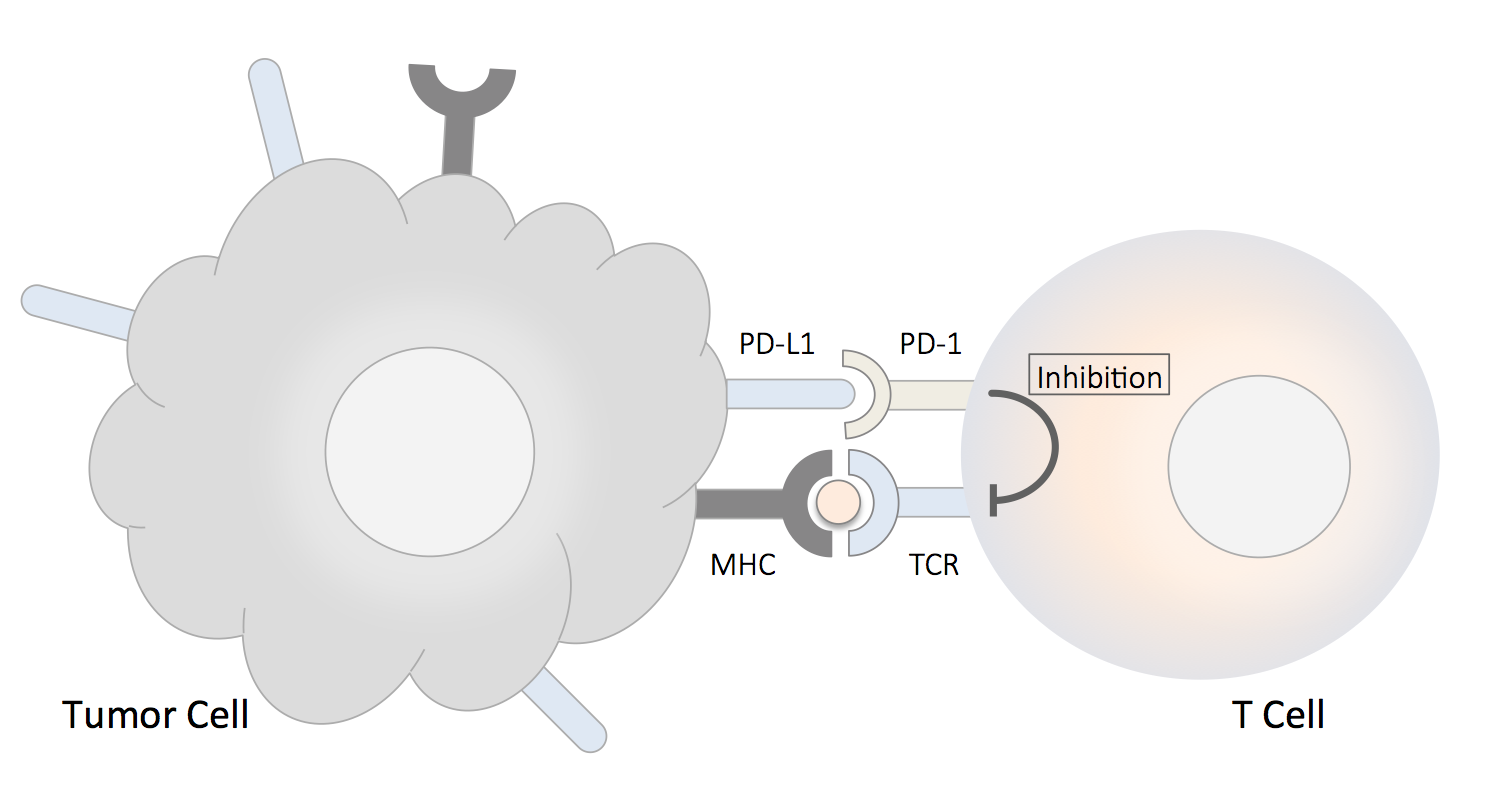
Introduction of the PD-1 and PD-L1 Pathway
The PD-1 (the programmed death-1) receptor (also known as CD279) and the programmed death ligand-1 (PD-L1) are immune checkpoint proteins. PD-1 is an inhibitory receptor expressed on the surface of activated T cells, its ligand PD-L1 is expressed on epithelial and endothelial cells, in addition to different types of immune cells usch as antigen-presenting cells (dendritic cells, macrophages, B cells). PD-L1 can be both constitutively expressed and upregulated by interferons present in an inflammatory condition such as a chronic tissue infection. PD-L1 is also expressed on the surface of tumor cells. The binding of PD-1 and PD-L1 transmits an inhibitory signal into the T cell, which reduces cytokine production, suppress T cells proliferation and halt them from targeting tumor cells. The immune checkpoint pathway has emerged as an improtant tumor-evasion mechanism; therefore, targeting either PD-1 or PD-L1 can stimnulate the immune sytem and enhance the efficiency of T cell-mediated clearance of tumor cells.
PD-1, PD-L1 and Cancer Therapy
Inhibitory receptor PD-1 expressed on tumor-specific T cells lead to compromised activation and suppressed effector functions such as proliferation, cytokine secretion, and tumor cell lysis when binds to PD-L1 expressed by tumor cells. Immune checkpoint blockade has gained momentum in cancer immunotherapy using monoclonal antibodies directed against PD-1 and PD-L1 [1]. Effective cancer therapies using this approach included advanced melanoma, non-small cell lung cancer (NSCLC), renal cell cancer (RCC) and other tumor types [2]. However, it has to be noted that most tissues rely on PD-L1 expression to limit T cell response, so the systemic administration of PD-L1/PD-1 blocking antibodies still carries the risk of breaking peripheral tolerance.
CRISPR and PD-1, PD-L1
CRISPR-Cas9 system has been developed for effective genome editing. Using a single-guide RNA (sgRNA), the Cas9 endonucleaes can be precisely guided to target sites to produce DNA double-strad breaks (DSBs), which initiate DNA repair processes that give rise to site-specific genomic modification. Since first applied in mammalian cells and animals in 2013 [3], CRISPR-Cas9-based therapeutic strategies has been actively implemented in the research of cancer modeling and treatment.
The CRISPR-Cas9 system can be directly applied to human cells by non-viral or viral delivery of genomic components encoding Cas9 and sgRNA [4]. Genetic deletion of PD-1 may be a useful method in enhancing efficiency of T-cell-based immunotherapy in cancers [5]. Knockout of PD-1 has been achieved using Cas9-sgRNA ribonucleoproteins and exogenous single-strand DNA templates to replace targeted nucleotides [6]. Similarly, it has been demonstrated that targeted gene knockout of PD-1 by sgRNA and Cas9 in human T cells resulted in a significant reduction of PD-1 expression, which up-regulated T cell immune responses and enhanced cytotoxicity on cancer cell lines [7]. A Jurkat T cell-based lentiviral CRISPR toolbox was developed to facilitate the research on T cell function [8].
Recently, a clinical trial has recently been approved by the US National Institutes of Health (NIH) Recombinant DNA Advisory Committee. In this clinical trial, PD-1, the endogenous T cell receptor and NY-ESO-1 receptor will be knocked out by CRISPR-Cas9 simultaneously. The first clinical trial of CRISPR/ Cas9 has been initiated [9]. Similar trials with PD-1-knockout by CRISPR-Cas9 in autologous T cells for prostate (NCT02867345), bladder cancer (NCT02863913), and renal cell carcinoma (NCT02867332) are also being initiated.
The CRISPR gene-edited CAR T cells have shown potent anti-tumor activities in vitro and in animal models [10]. PD-L1 expression on tumor cells can render human CAR T cells hypo-functional, resulting in impaired tumor clearance in a subcutaneous xenograft model. Disruption of PD-1 by CRISPR/Cas9 in anti-CD19 CAR T cells augmented CAR T cell mediated killing of tumor cells in vitro and enhanced clearance of PD-L1+ tumor xenografts in vivo [11].
References
- Keir, M. E., Butte M. J., Freeman G. J. & Sharpe A. H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008).
- Suzanne, L. T. et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 339(6121):819-23 (2013).
- H. Yin, C.-Q. Song, J.R. Dorkin, L.J. Zhu, Y. Li, Q. Wu, A. Park, J. Yang, S. Suresh, A. Bizhanova, Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo, Nat. Biotechnol. 34, 328-333 (2016).
- S.L. Topalian, C.G. Drake, D.M. Pardoll, Immune checkpoint blockade: a common denominator approach to cancer therapy, Cancer Cell, 27, 450-461 (2015).
- K. Schumann, S. Lin, E. Boyer, D.R. Simeonov, M. Subramaniam, R.E. Gate, G.E. Haliburton, J.Y. Chun, J.A. Bluestone, J.A. Doudna, Generation of knock-in primary human T cells using Cas9 ribonucleoproteins, Proc. Natl. Acad. Sci. U S A. 112, 10437-10442 (2015).
- S. Su, B. Hu, J. Shao, B. Shen, J. Du, Y. Du, J. Zhou, L. Yu, L. Zhang, F. Chen, CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients, Sci. Rep. 6, 20070 (2016).
- Chi S, Weiss A, Wang H. A CRISPR-Based Toolbox for Studying T Cell Signal Transduction. Biomed Res Int. 2016:5052369 (2016).
- Cyranoski D, CRISPR gene-editing tested in a person for the first time. Nature 539:479 (2016).
- Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res. 23(9):2255-2266 (2017).
- Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 7(1):737 (2017).