 |
Ed Davis, Ph.D. |
Genome Editing-the ability to make specific changes at targeted genomic sites-is of fundamental importance in biology and medicine (for reviews, see Bogdanove & Voytas, 2011; van der Oost, et al.,2013). Two genome editing technologies have emerged recently that exploit bacterial systems for plant pathogenesis or adaptive immunity: TALEN (Transcription Activator-Like Effector Nucleases) and CRISPR (Clustered, Regularly Interspaced, Short Palindromic Repeats), respectively. Both TALEN and CRISPR use endonucleases that initiate double-strand breaks (DSBs) at virtually any genomic target sequence, and can be used for many applications, including gene knock out, transgene knock in, gene tagging, and correction of genetic defects. However, researchers are often unaware of some of the work required to identify their desired modification in their cell lines. In this Technical Note, we discuss what you need to do for genome editing in mammalian cell culture after you have obtained your reagents from GeneCopoeia, the so-called “Downstream work”.
Upon receipt of plasmids
We recommend a specific workflow for how to use your GeneCopoeia genome editing plasmids (Figure 1). First, transform E. coli and make a frozen stock, so you’ll have an easily-renewable supply of DNA.
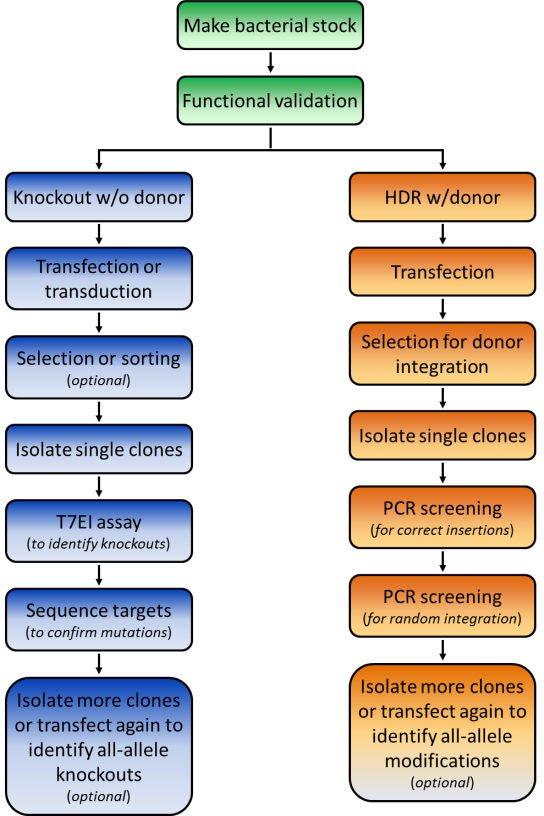
Figure 1. Pathways for repair of DSBs induced by genome editing tools. Left: Non-homologous end joining. Right: HR in the presence of a donor template.
|
Our plasmids are Ampicillin-resistant and can transform E. coli using standard protocols. Next, prepare a large amount of transfection-ready DNA using a commercial kit. Functional validationBefore starting your experiments we recommend functional validation. This is optional, but will give you a good idea of how well your TALENs or CRISPRs function. We also offer functional validation in human HEK293T and mouse Neuro2a cells. You can also carry out the validation assays yourself in your chosen cell line. For functional validation, we recommend using the mismatch cleavage assay (Qiu, et al., 2004). with T7 Endonuclease I. This assay looks for the presence of insertion or deletion (“indel”) mutations resulting from nonhomologous end joining (NHEJ) of DSBs at the target site. The basic steps of this assay are outlined in Figure 2A. Briefly, cells are transiently transfected using standard methods, or transduced if using lentivral particles. Next, cells are harvested and used to prepare genomic DNA,followed by PCR using primers flanking the DSB site.
|
Finally, the PCR products are denatured, re-annealed, and digested with T7 Endonuclease I, which cleaves double stranded DNA containing mismatches. If cleavage was successful, then an agarose gel of the reaction products will show the presence of three bands: The full-length, undigested homoduplex band, and two smaller bands that are mismatch cleavage products (Figure 2B).
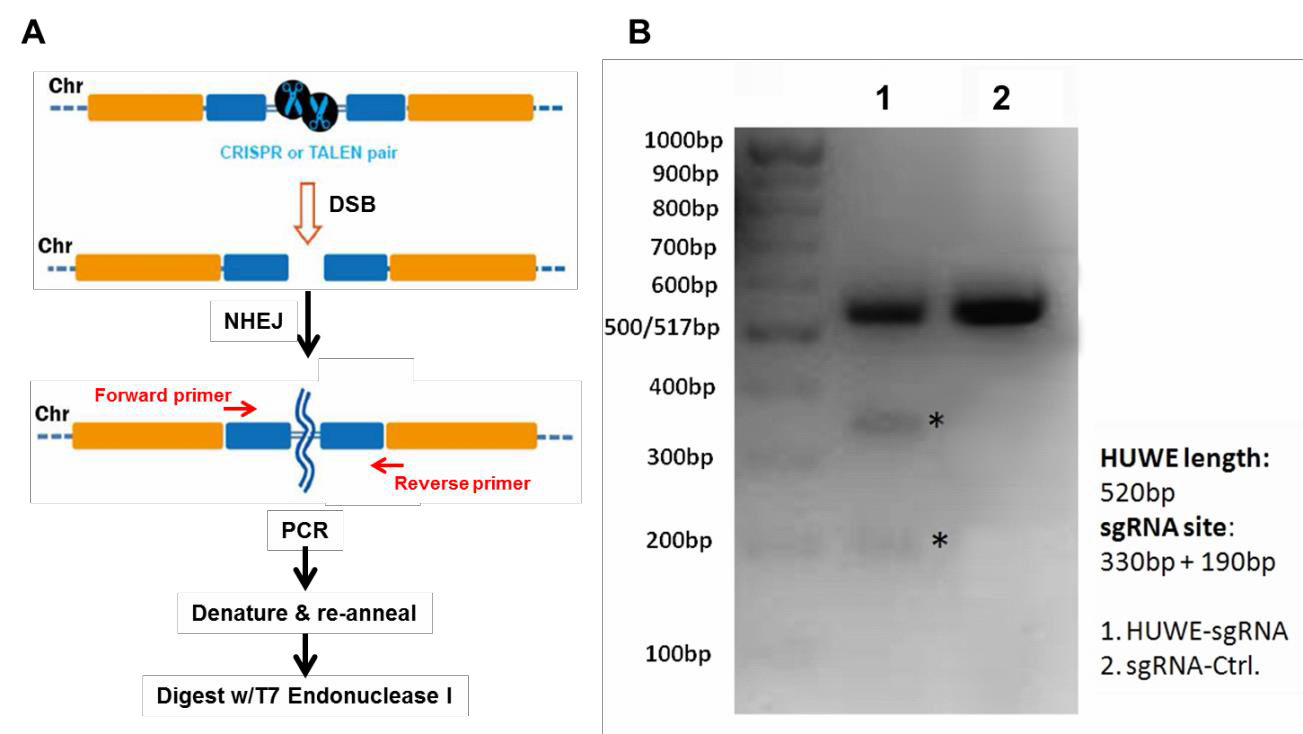 |
Figure 2. Mismatch cleavage assay. A. General scheme beginning with DSB formation, through digestion of mismatched PCR products with T7 Endonuclease I. B. Agarose gel of T7 endonuclease I digestion products. Lane 1 shows undigested as well as digested products, indicating successful cleavage. Lane 2 shows the reaction from a transfection using a scrambled negative control sgRNA.
Transfection or Transduction
Our non-viral plasmids for TALEN and CRISPR can be transfected by standard methodologies GeneCopoeia offers our EndofectinTM brand of transfection reagents. Alternatively, you can use whichever method works best for your cells. Our lentiviral CRISPR plasmids are compatible with 3rd- generation lentiviral packaging reagents. We also offer our own brand of LentifectTM lentiviral packaging reagents, as well as lentiviral packaging services.
Both our non-viral as well as the lentiviral plasmids typically carry drug selection markers and fluorescent reporter genes, to aid you in your transfection and transduction procedures (Figure 3).
Clonal isolation
In genetics, it is essential to purify mutants away from a population-a practice known as clonal isolation. Clonal isolation minimizes the potential effects of unwanted modifications resulting from cell division or off-targeting, which could confound your results. When you are ready to begin screening, you’ll either need to plate cells such that they will form individual colonies, which you can then pick off the dish, or do serial dilutions in 96-well plates.
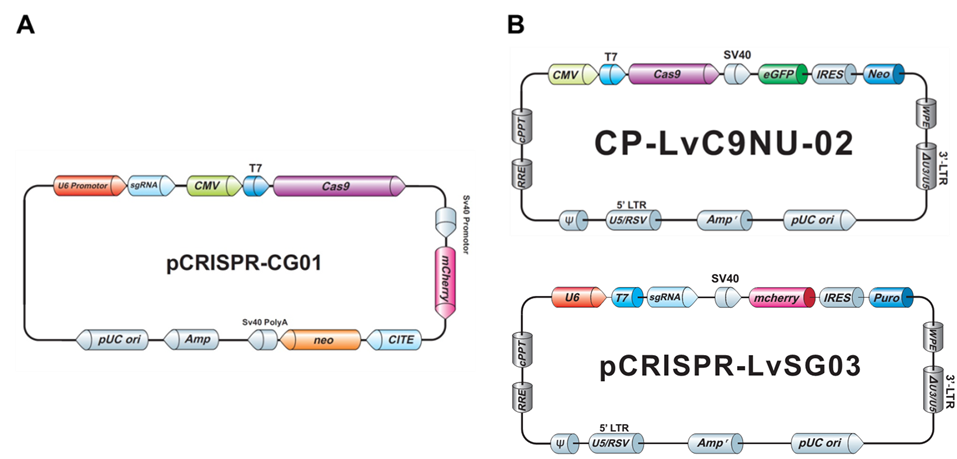 |
Figure 3. Examples of GeneCopoeia genome editing plasmids. A. Non-viral all-in-one plasmid containing CRISPR Cas9 nuclease, one CRISPR sgRNA, the neo gene for G418 selection, and mCherry as a fluorescent reporter. B. Lentiviral plasmids. Top, CRISPR Cas9 nuclease vector with the neo gene and eGFP. Bottom, CRISPR sgRNA plasmid with the puro gene for puromycin selection and mCherry.
Screening
The type of screening required depends on the type of genomic modification you are making. Three common situations are discussed:
1. Knockout by NHEJ
The fastest way to screen for NHEJ-mediated knockouts is by using the mismatch cleavage assay, as described above for functional validation. Once modified clones are identified, you’ll need to perform Sanger sequencing on the PCR products, to determine the exact nature of the mutation.
2. HR applications using a donor plasmid
The frequency of homologous recombination mediated by TALEN or CRISPR is typically much less than that of NHEJ, so it might seem you’d need to screen even more colonies to identify a modification with a donor plasmid. However, a donor plasmid provides drug selection, and, if present, a fluorescent reporter. GeneCopoeia donor plasmids carry drug selection and fluorescent reporter genes (Figure 4). After transfection, cells are plated in the presence of a drug, like puromycin, at low density to allow formation of single colonies. Alternatively, serial dilutions can be done in 96 well plates. Colonies that grow in the presence of the drug are candidates for stable cell lines carrying the modification.
Once drug resistant and/or fluorescent reporter-expressing clones are identified, the screening procedure differs dramatically from that used for NHEJ. For knockouts, the selection/reporter cassette is typically integrated in the protein-coding region, sometimes with a small deletion at the site of cleavage by TALEN or CRISPR. In other cases, a defined chromosomal region of DNA might be deleted and replaced with the cassette, or a gene might carry an in-frame fusion tag.
For screening, PCR with primers spanning the junction between the unmodified chromosome sequence and the sequence being inserted should be used. On both the left and right side of the insertion, one primer is designed for the homology arm, for synthesis in the direction of the marker, while the opposite primer targets the insertion for synthesis toward the homology arm. If the integration was successful, you will then be able to visualize bands of the expected sizes on an agarose gel.
The fastest way to screen for NHEJ-mediated knockouts is by using the mismatch cleavage assay, as described above for functional validation. Once modified clones are identified, you’ll need to perform Sanger sequencing on the PCR products, to determine the exact nature of the mutation.
The frequency of homologous recombination mediated by TALEN or CRISPR is typically much less than that of NHEJ, so it might seem you’d need to screen even more colonies to identify a modification with a donor plasmid. However, a donor plasmid provides drug selection, and, if present, a fluorescent reporter. GeneCopoeia donor plasmids carry drug selection and fluorescent reporter genes (Figure 4). After transfection, cells are plated in the presence of a drug, like puromycin, at low density to allow formation of single colonies. Alternatively, serial dilutions can be done in 96 well plates. Colonies that grow in the presence of the drug are candidates for stable cell lines carrying the modification.
Once drug resistant and/or fluorescent reporter-expressing clones are identified, the screening procedure differs dramatically from that used for NHEJ. For knockouts, the selection/reporter cassette is typically integrated in the protein-coding region, sometimes with a small deletion at the site of cleavage by TALEN or CRISPR. In other cases, a defined chromosomal region of DNA might be deleted and replaced with the cassette, or a gene might carry an in-frame fusion tag.
For screening, PCR with primers spanning the junction between the unmodified chromosome sequence and the sequence being inserted should be used. On both the left and right side of the insertion, one primer is designed for the homology arm, for synthesis in the direction of the marker, while the opposite primer targets the insertion for synthesis toward the homology arm. If the integration was successful, you will then be able to visualize bands of the expected sizes on an agarose gel.

Figure 4. Example of a GeneCopoeia donor plasmid for HR applications.
Using drug selection can lead to random integration of the donor plasmid, which must be ruled out. GeneCopoeia donor vectors carry the herpes simplex virus (HSV) thymidine kinase (TK) gene outside the region of recombination (Figure 4). If ganciclovir is included in the selection medium, then cells containing random integration of the donor plasmid will usually die, leaving a population enriched for those containing correct donor integration. Less frequently, random integration can exclude the TK gene, so PCR and/or Southern blotting should still be employed to rule out such events.
Finally, a gene modified by HR needs to be sequenced, so that both the chromosome structure surrounding the modified site is verified, and to detect the presence of an introduced mutation, if applicable.
At GeneCopoeia, our Genome Editing team has a wealth of expertise and experience in the design, construction, and use of TALEN and CRISPR reagents in mammalian systems. We start at TALEN and CRISPR design, delivering you sequence-verified plasmid DNA. We also offer functional validation services for your TALEN and CRISPR constructs. Further, we can construct stable cell lines or transgenic mice containing your TALEN- or CRISPR-mediated modification of interest. We also offer scientific consulting services to help you plan your genome editing projects. For more information, visit our website: https://www.genecopoeia.com. You can also call us at 1-301-762-0888, or email inquiry@genecopoeia.com
Things to look out for
Before devising your transfection and screening strategy, you should consider several factors that will affect the number of colonies you need to screen and, if necessary, the number of rounds of transfections needed to be carried out:
- Transfection efficiency. The percentage of cells that receive the TALEN or CRISPR reagents is critical for determining the number of colonies that need to be screened. If the transfection efficiency of your cell line is 40%, then you should expect to screen twice as many colonies as you would in a cell line with 80% transfection efficiency. If your cell line’s transfection efficiency is low, you can enrich for transfected cells by sorting using a fluorescent marker on one of the plasmids. You can also use drug selection for a short time period. We recommend estimating the transfection efficiency of your cell line using a plasmid that constitutively expresses a fluorescent reporter gene such as GFP.
- Copy number. For complete knockouts or mutagenesis, you might need to modify all alleles of the gene. Many people assume that they need to modify two alleles in a diploid cell line. However, many popular immortalized cell lines, such as HeLa, are aneuploid or polyploid, and so could contain more than 2 copies of your gene. The efficiency of modifying a single allele is higher than modifying multiple alleles in one cell. Therefore, if you need more than two alleles modified, you’ll need to screen more colonies than you would for a double allele modification. You might need to perform multiple rounds of transfection.
- Cutting efficiency. The efficiencies of indel formation by TALEN and CRISPR typically range from approximately 5% to about 70%. As with transfection efficiency, you should expect to screen twice as many colonies when the cutting efficiency is 20% as you would when the cutting efficiency is 40%.
The combination of factors will influence the number of colonies you’ll need to screen in order to find one with your modification. Simplistically, if you want a double-allele knockout in a diploid cell line using a CRISPR with 50% cutting efficiency and your transfection efficiency is 60%, then the percentage of colonies with a single allele modified would be 0.5 x 0.6 = 30%. For double allele modification, the percentage of positive colonies would be 15%. In this scenario, you’d need to screen at least 20 colonies to find one containing a double allele modification. If the transfection and cutting efficiencies are even lower, and your cell line contains more than two copies of your target, you will need to screen many more colonies in order to find one with a complete knockout.
References
Bogdanove & Voytas (2011). TAL Effectors: Customizable proteins for DNA targeting. Science 333, 1843.
Qiu, et al. (2004). Mutation detection using Surveyor™ nuclease. Biotechniques 36, 702.
van der Oost, et al. (2013). New Tool for Genome Surgery. Science 339, 768.
| Copyright ©2014 GeneCopoeia, Inc. www.genecopoeia.com TNGE3-070914 |