General
Answer: If you are doing simple gene knockouts in humans, mice, or rats, you can order TALENs or CRISPR sgRNAs on our website. All you need to do is go to the
TALEN/CRISPR search page, search for your gene, and then choose the appropriate clones that will work for your system. You can also order donor clones for these knockouts from the search results page. Please note that these TALENs and CRISPR sgRNAs are designed by default to target the protein coding-region as closely as possible to the initiator ATG of the splice variant (accession number) you select. This design strategy does not consider other possible variants of the gene. If the gene has multiple variants and you would like to target one particular exon, one unique variant, multiple variants, or all variants, or if you are doing a different application, such as introducing a point mutation, then you will need to
contact us and, after determining what you need, we will send you a custom quote.
Answer: For sgRNA clones (including both all-in-one Cas9/sgRNA clones and sgRNA-only clones) as well as TALE clones, the default delivery format is bacterial stock. You have the option of ordering purified DNA for these clones for an additional charge. For
HDR donor clones, the default delivery format is purified DNA. In addition, we offer
ready-to-use lentiviral particles for Cas9 and sgRNA delivery.
Answer: The turnaround time for sgRNA clones (including both all-in-one Cas9/sgRNA clones and sgRNA-only clones) and TALE clones is 2-3 weeks. The turnaround time for
HDR donor clones depends greatly on the nature of the modification that the clone is being used for. For HDR donor clones used for simple knockout, the turnaround time is 2-4 weeks. Other HDR donor clones, such as those used for fusion tagging or mutagenesis, can take 6-8 weeks, but can also take longer.
Answer: Yes. We sequence the inserts of each CRISPR sgRNA or TALE clone, and provide you with datasheets that show the full sequence of each clone (including HDR donor clones), a map, restriction enzyme digestions sites, and suggested sequencing primers. To obtain these datasheets, you just need to visit our
datasheet download page on our website. You will need an account on our website, your catalog number(s), and your sales order number.
Answer: In the presence of drug, the only way for cells to survive is to integrate the plasmid into the chromosome, so it is possible to get drug-resistant clones that were transfected only with the donor plasmid. However, such integration is random. CRISPR increases donor targeting frequency by several orders of magnitude, and CRISPR-mediated donor integration is usually targeted.
Answer: Yes. For this you will need a donor that is homologous to your locus on either side of the base you want to mutate. The donor can be either a plasmid or a single-strand oligonucleotide. The donor is co-transfected with the genome editing tool (TALEN or CRISPR). Generation of a double strand break (DSB) leads to repair of the break by homology-directed repair (HDR) using the donor as a template.
GeneCopoeia recommends our donor plasmid design and construction service. We will construct a donor plasmid that contains a defined modification, flanked by a selectable marker such as puromycin resistance, and homologous arms from your target region. The donor may or may not also include a fluorescent reporter such as GFP. The markers can be flanked by loxP sites, to permit Cre-mediated removal, if desired. Use of a GeneCopoeia-designed donor plasmid allows you to select for edited clones and reduces the number of clones required for screening. You can also purchase our donor cloning vectors for do-it-yourself donor clone construction.
Answer: There are two strong reasons to use HDR for genome editing: 1) It is precise and controllable. Any desired change can be implemented; 2) You will be able to use a knocked-in marker (such as drug resistance or fluorescence), which will greatly facilitate your ability to identify candidate clones that have the modification you want. In addition, some applications, such as fusion tagging, require using HDR.
GeneCopoeia recommends our donor plasmid design and construction service. We will construct a donor plasmid that contains a defined modification, flanked by a selectable marker such as puromycin resistance, and homologous arms from your target region. The donor may or may not also include a fluorescent reporter such as GFP. The markers can be flanked by loxP sites, to permit Cre-mediated removal, if desired. Use of a GeneCopoeia-designed donor plasmid allows you to select for edited clones and reduces the number of clones required for screening. You can also purchase our donor cloning vectors for do-it-yourself donor clone construction.
Answer: Our genome editing products can be used for virtually all species. Our standard plasmids for both TALE systems and CRISPR are designed for work in mammalian cells. In addition, these plasmids can be used as templates for T7 promoter-driven in vitro transcription, for introduction into mice, zebrafish, Drosophila, and many other model organisms. Further, we can generate custom constructs that can be used in a wide variety of organisms.
Answer: Yes. The donor must be present when the double strand break (DSB) is generated in order to be used as a repair template. Otherwise, the cell must use non-homologous end joining (NHEJ) to repair the DSB, because unrepaired DSBs are lethal.
Answer: Our TALE and CRISPR plasmids typically only integrate into the host genome at very low frequency in transfection experiments. However, after clonal selection for edited cells, we recommend screening clones for those which have lost the plasmids. This can be done by testing clones to see if they have become sensitive to the antibiotic of the resistance gene on the plasmid, or if they no longer express the plasmid’s fluorescent marker (where applicable). Our lentiviral clones, when packaged into lentiviral particles and used to infect cells, are expected to integrate randomly into chromosomes.
Answer: The presence of the CMV or other promoters driving expression of the Cas9 or TALE coding sequences permits expression from the plasmid DNA. sgRNA transcription is driven by the U6 promoter. We recommend that you use the most efficient method for your cell type of interest. The following are all acceptable approaches for delivering our genome editing tools into cells:
1. Standard transfection. GeneCopoeia recommends our EndoFectin™ transfection reagents. Transfection can be used for either DNA, RNA, or Cas9 nuclease protein.
2. Electroporation, which can be used for either DNA, RNA, or Cas9 nuclease protein.
3. Lentiviral transduction (cannot be used with HDR donors).
4. Micro-injection of either DNA, mRNA, .or Cas9 nuclease protein. The plasmids can be used as templates for in vitro transcription of RNA, or synthetic RNA can be used.
Answer: If you are using TALEN or CRISPR to create mutations in your gene of interest without the use of a homologous donor, then you will need to undertake much time-consuming, labor-intensive downstream work in order to identify edited clones. After transfection, you will need to isolate many colonies grown up from single cells. Next, the screening procedure depends on the nature of the modification, as described below:
1. If you are making an insertion or deletion, the easiest way to screen your cells is by PCR using primers flanking the modified site, provided that the insertion or deletion is large enough to detect by standard agarose gel electrophoresis.
2. For very small insertions or deletions, you can screen your clones using GeneCopoeia’s IndelCheck™ T7 endonuclease I assay, which is a method that detects mutations by cleaving double stranded DNA containing a mismatch. You can also screen using Sanger sequencing of PCR products. Ultimately, Sanger sequencing needs to be done to verify the presence of the modification.
3. If you are introducing a point mutation, then you can use either real-time PCR or Sanger sequencing to detect the modification.
4. If the modification you are introducing creates or destroys a restriction enzyme site, then enzyme cleavage of PCR products can be used to distinguish between modified and unmodified alleles.
5. Finally, either Sanger sequencing of PCR products or Next Generation sequencing of whole genomes can be used to screen for modifications. Regardless of which screening method you choose, it is also important that you are able to determine whether only a portion or all of the alleles have been modified.
In order to reduce the amount of time and effort required to identify edited clones, GeneCopoeia recommends our donor plasmid design and construction service. We will construct a donor plasmid that contains a defined modification, flanked by a selectable marker such as puromycin resistance, and homologous arms from your target region. The donor may or may not also include a fluorescent reporter such as GFP. The markers can be flanked by loxP sites, to permit Cre-mediated removal, if desired. Use of a GeneCopoeia-designed donor plasmid allows you to select for edited clones and reduces the number of clones required for screening. You can also purchase our donor cloning vectors for do-it-yourself donor clone construction.
Answer: For safe harbor integration, we recommend PCR using primers designed to amplify the recombination loci, or Southern blotting, which can rule out random integration. All GeneCopoeia
Safe Harbor Gene knock-in Kits contain PCR primer pairs intended for this exact purpose. For other editing experiments using HDR, junction PCR can be be used to identify edited sites. The junction PCR method will will only yield product if donor integration occurred at the correct genomic site. Southern blotting or PCR using primers recognizing the plasmid backbone can also be used to rule out random integration.
Answer: Yes. Even though frameshifts are not possible with miRNAs and other noncoding RNAs, an indel occurring in a critical region, such as the mature sequence of a miRNA, should be enough to abolish its function.
Answer: The vector backbones of our TAL effectors or CRISPR sgRNAs are designed to not replicate in the host. These plasmids, which are transiently transfected, will typically be lost after several rounds of cell division and will not further affect the host cell. After transfection, cells are plated at low density to promote the formation of single colonies. These colonies should be screened to ensure that they have lost the plasmid(s). This can be done by testing clones to see if they have become sensitive to the antibiotic of the resistance gene on the plasmid, or if they no longer express the plasmid’s fluorescent marker (where applicable). However, even if the TALEN or CRISPR plasmid integrates, it can no longer cut the site after it is edited, because NHEJ destroys the TALEN or sgRNA recognition site. To be completely assured that the transfection is transient, we recommend delivering RNA or
Cas9 nuclease protein instead of plasmid DNA. If you are using HDR, we recommend engineering synonymous mutations into the donor to destroy the TALEN or sgRNA recognition site without changing the amino acid sequence.
Answer: Yes. Both TALEN and CRISPR have been shown to be able to disrupt multiple copies at once. The efficiency varies depending on different factors, such as cell type, transfection efficiency and TALEN/CRISPR activity.
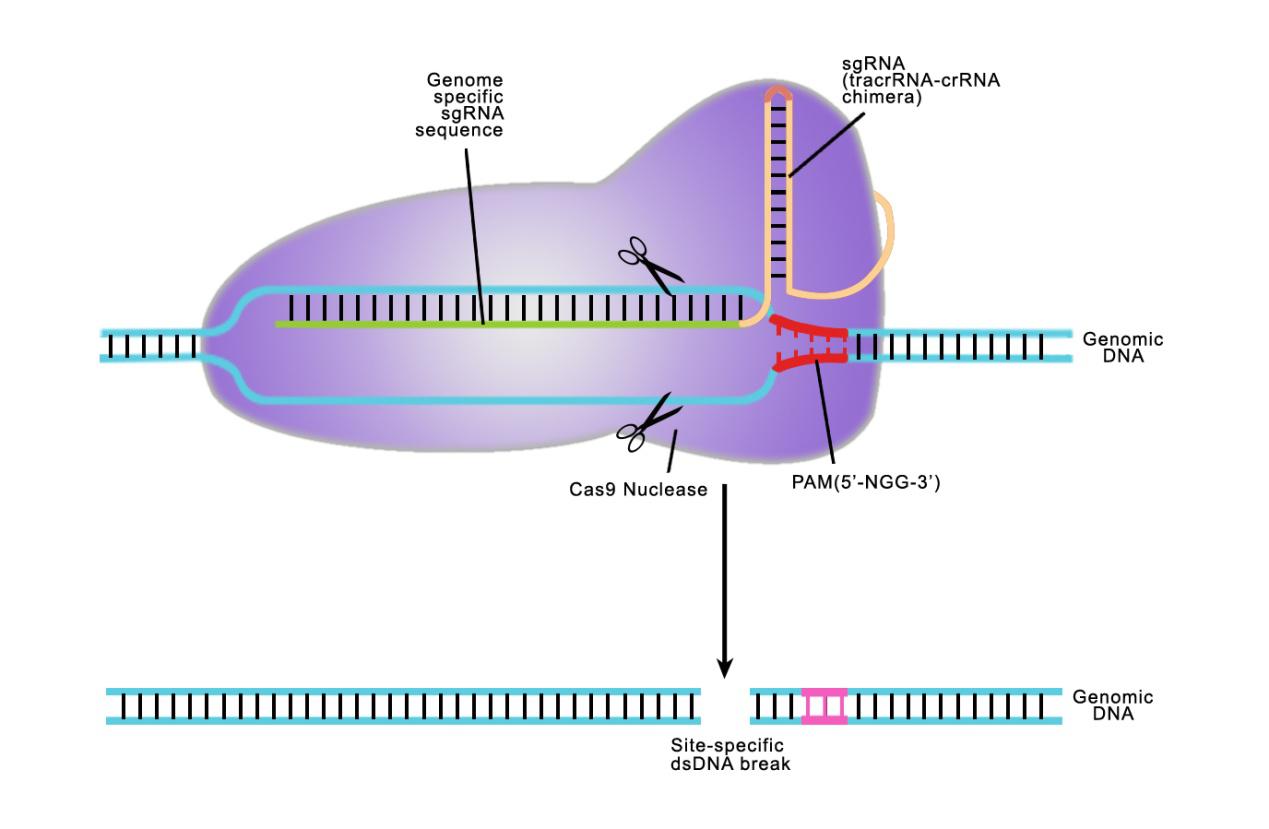
Answer: Both TALE and CRISPR recognize a specifi target sequence to initiate genome editing. The biggest difference is in how they do it. CRISPR uses a single guide RNA (sgRNA) homologous to a 20 nucleotide target sequence. This target sequence must be immediately followed by a 3 nucleotide N-G-G sequence known as the Protospacer Adjacent Motif (PAM). The single guide RNA guides the Cas9 nuclease to the desired site. When using the CRISPR system, we need to design the sequence of sgRNAs.
TALEs are proteins that recognize target sequences using variable amino acids in a series of 34 amino acid repeats. These variant amino acids are known as Repeat Variable Diresidues (RVDs). Each repeat contains one RVD, and each of the RVDs recognizes a specific nucleotide.
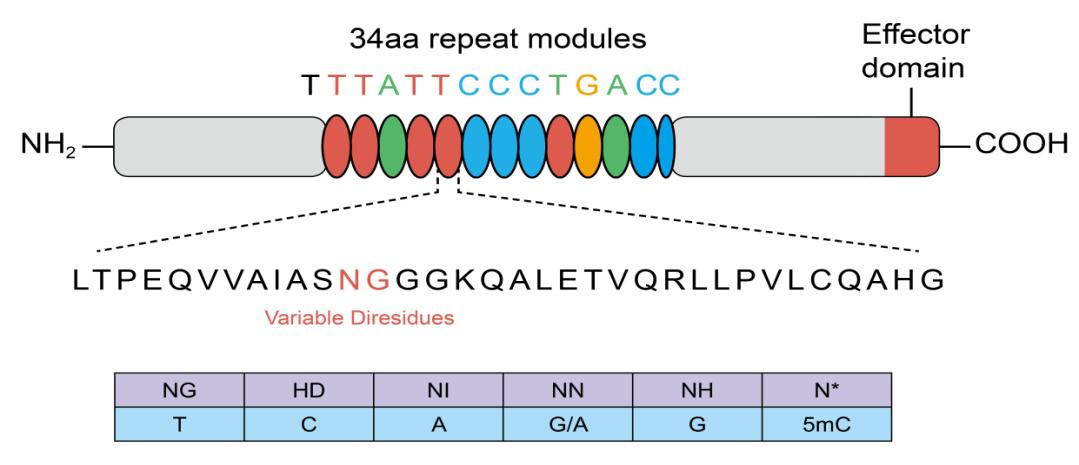
TALEs are fused to their genome editing motifs, which can include nucleases or transcriptional modulators. When using TALE technology systems, we need to assemble RVDs in the correct order to match the target sequence.
Answer: There is never an easy answer to this question. It all depends on what you are trying to accomplish. Each system has its advantages and disadvantages. CRISPR tends to edit genes at higher efficiency than TALEN. Also, CRISPR is not sensitive to DNA methylation, and is much more amenable to multiplexing, giving it further advantages over TALEN. On the other hand, CRISPR, including the Cas9 nickase, is more prone to modifying off-target sites, which, depending on your particular application may or may not be an issue. There are also “high-fidelity” variants of Cas9 nuclease that edit genes with greater specificity, but sometimes with reduced efficacy and with increased design constraints.
TALE
Answer: Yes.
CRISPR
Answer: Yes. We have the reagents for the Cas9 D10A nickase, and have successfully tested our double nickase designs. However, in order to create mutagenic DSBs, the nickase requires the correct targeting of two appropriately-spaced sgRNAs on opposite strands, flanking the break site. Because proper sgRNA targeting requires the presence of the N-G-G “PAM” site immediately following the recognition site, it might not always be possible to use the nickase for DSB formation. There are also “high-fidelity” variants of Cas9 nuclease that edit genes with greater specificity than wild type Cas9, but sometimes with reduced efficacy and with increased design constraints. However, since these high fidelity variants use only one sgRNA, they are easier to work with than Cas9 nickases. In addition, using
Cas9 nuclease protein instead of a DNA plasmid could help reduce off-target activity.
Answer: Yes. To create a DSB, the nickase requires the correct targeting of two appropriately-spaced sgRNAs on opposite strands, flanking the break site. This is sufficient to stimulate HDR between the target site and the donor. While this method has the advantage of potentially fewer off-target NHEJ-mediated mutations, since single strand nicks are repaired with higher fidelity than DSBs, it is not without limitations. Proper sgRNA targeting requires the presence of the N-G-G “PAM” site immediately following the recognition site. Therefore, it might not always be possible to use the nickase for HDR. There are also “high-fidelity” variants of Cas9 nuclease that edit genes with greater specificity than wild type Cas9, but sometimes with reduced efficacy and with increased design constraints. However, since these high fidelity variants use only one sgRNA, they are easier to work with than Cas9 nickases. In addition, using
Cas9 nuclease protein instead of a DNA plasmid could help reduce off-target activity.
Answer: We only sell plasmids containing our custom-designed CRISPR sgRNAs. If you need a negative control, we also sell CRISPR plasmids containing a scrambled sgRNA.
Answer: Yes.
Answer: Yes. There is a double mutant of the Cas9 nuclease that completely abolishes nuclease activity. This mutant can be fused to a transcriptional modulator such as VP64 and targeted to specific genes. You can also use the catalytically dead Cas9 with properly-designed sgRNAs to repress, or interfere with, gene expression. For more information, please visit our pages on
CRISPR activation (CRISPRa) and
CRISPR interference (CRISPRi).
Answer: Unfortunately, no. Lentiviruses enter cells as RNA, but HDR donors must enter the cells as DNA at the same time as Cas9 and the sgRNAs.
Answer: Cas9 stable cell lines are primarily useful for lentiviral CRISPR delivery and CRISPR sgRNA library screening. sgRNA library screening is typically done with lentivral delivery. In principle, it is possible to either co-infect your cell line with one Cas9-expressing lentivirus and with an sgRNA-expressing lentivirus, or with a single “all-in-one” lentivirus (Cas9 and sgRNA expressed in the same virus). However, in practice, due to the large size of the Cas9 gene (4.4 kb), the titers of Cas9-expressing lentiviruses tend to be much lower than sgRNA-expressing lentiviruses. Therefore, we recommend using a cell line that has previously had Cas9 stably integrated in the genome for infection with lentiviral-based sgRNA libraries. GeneCopoeia carries a large collection of
pre-made Cas9-expressing stable cell lines, as well as stable cell lines expressing the machinery for
CRISPR activation (CRISPRa) and
CRISPR interference (CRISPRi). You can also purchase reagents that you can use to build a Cas9 stable cell line yourself, or you can have GeneCopoeia create a custom Cas9-expressing stable cell line as a
custom cell service project.
sgRNA libraries
Answer: Lentiviral particles, transfection-ready DNA, and bacterial stock.
Answer: Yes. Our
sgRNA libraries are available standard as pools. However, if you wish to receive your libraries as individually arrayed clones, simply
contact us for a custom quote.
Answer: Yes. However, we strongly recommend that you use a cell line that is stably expressing Cas9 to transduce sgRNA-expressing lentiviral particles with, in order to best maintain consistent representation of each sgRNA in the library. To save you time and effort, GeneCopoeia provides a large collection of
Cas9-expressing stable cell lines that are ideally suited for sgRNA library screening. If you would like to create a Cas9-expressing stable cell line yourself, you should first transduce your cells with Cas9-expressing lentiviral particles. Once you have established the stable Cas9-expressing cell line, use the sgRNA libraries for transduction.
Answer: Yes. The lentiviral plasmids are “dual-use”, so that they can either be packaged into lentiviral particles or transfected into cells by standard transfection methods.
Answer: Our sgRNA representation does not need to be validated by Next Generation Sequencing. Each library is small compared with the genome-wide libraries, and each sgRNA clone is constructed individually, cultured in E. coli individually, then pooled as E. coli in approximately equal amounts. From those pools we prepare DNA and then, if necessary, lentiviral particles.