by admin | Aug 28, 2014 | Resources
 |
|
Ed Davis, Ph.D. |
Introduction
Genome Editing-the ability to make specific changes at targeted genomic sites-is of fundamental importance to researchers in biology and medicine (Bogdanove & Voytas, 2011; van der Oost, et al.,2013). Two genome editing technologies have emerged recently that exploit bacterial systems for plant pathogenesis or adaptive immunity: TALEN (Transcription Activator-Like Effector Nucleases) and CRISPR (Clustered, Regularly Interspaced, Short Palindromic Repeats), respectively. Both TALEN and CRISPR use endonucleases that initiate double-strand breaks (DSBs) at virtually any genomic target sequence, and are used for many applications, including gene knockout, transgene knock-in, gene tagging, and correction of genetic defects. While both technologies are popular, the decision to choose one technology over the other is not always clear, and GeneCopoeia customers occasionally ask us for advice. In this Technical Note, we compare the advantages and disadvantages of TALEN and CRISPR, with the goal of arming customers with enough information to choose which technology to go with when ordering their reagents from us.
Genome editing
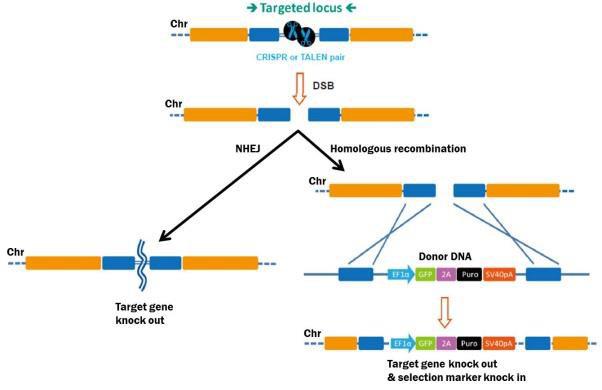
Genome editing starts with efficient DSB generation in the target DNA (Figure 1). DSBs are repaired either by homologous recombination (HR), or, in the absence of a homologous repair template, via non-homologous end joining (NHEJ). NHEJ causes small insertions or deletions as the broken ends are pieced back together. This proclivity for indel generation is exploited as a convenient method for knocking genes out. Both TALEN, which is comprised of a pair of DNA binding proteins fused to the FokI nuclease, and CRISPR, which is a complex between the Cas9 nuclease and a target-specific single guide RNA (sgRNA), can edit DNA through either HR or NHEJ.

Figure 1. Pathways for repair of DSBs induced by genome editing tools. Left: Non-homologous end joining. Right: Homologous recombination in the presence of a donor template.
Comparisons between TALEN and CRISPR
There are four main concerns to think about when trying to decide between TALEN and CRISPR:
1. Specificity
Both TALEN or CRISPR provide high target site specificity, enabling researchers to make precise genetic alterations. CRISPR achieves this specificity through the sgRNA, which is an artificial fusion of two naturally occurring short RNAs (Jinek, et al. 2012). The sgRNA directs the S. pyogenes Cas9 nuclease to a 20 nucleotide target site on the chromosome, which must be immediately followed by an N-G-G trinucleotide known as the Protospacer Adjacent Motif, or PAM (Figure 2). The sgRNA hybridizes with the strand opposite the PAM site, and Cas9 nuclease cuts the DNA. Many recent papers have demonstrated that CRISPR can cause DSB formation at very high frequencies at the intended target site.
However, sgRNAs can tolerate, to varying degrees, up to five mismatches (non-Watson-Crick base pairing) with unwanted target sites (Fu, et al., 2013), and CRISPR has been shown to physically associate with many off-target sites in the genome (Kuscu, et al, 2014). In addition, some studies report very high levels of indel formation at unintended sites (Fu, et al. 2013), although others suggest that the initial estimates of off- target mutagenesis may have been overstated (Li, et al.,2013; Yang, et al., 2013; Wang, et al., 2014).
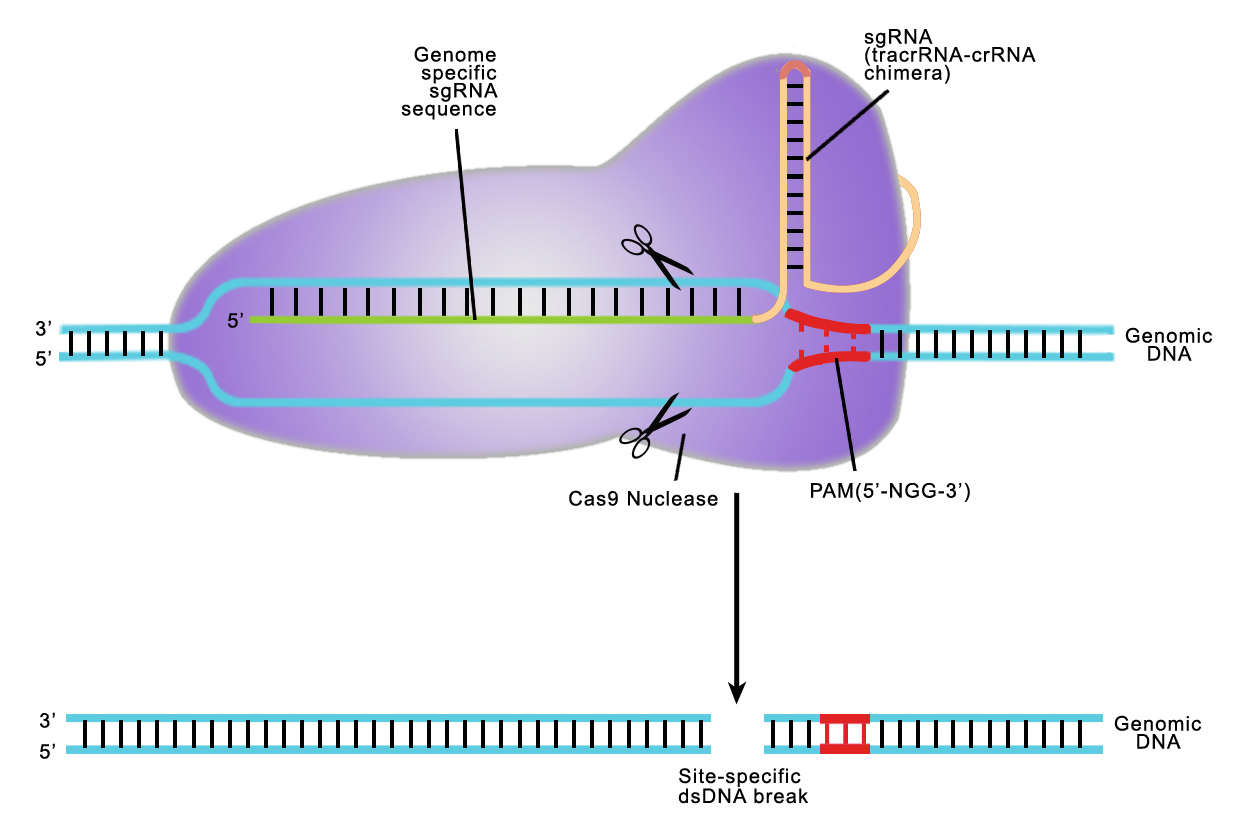
Figure 2. CRISPR-Cas9 target recognition.
Recent developments have significantly improved CRISPR specificity. One strategy employs paired single-strand break (“nickase”) mutants, which require targeting two sgRNAs on opposite strands flanking the target site (Mali, et al., 2013; Ran, et al., 2013). Individual nickases are still recruited to off- target sites, but nicks are less mutagenic than DSBs, so paired nickases dramatically reduce off-targeting. Further, shortening sgRNAs to as few as 17 nucleotides also reduces off-targeting (Fu, et al., 2014). Finally, a fusion between an inactive Cas9 and the FokI endonuclease (RNA-guided FokI nucleases, or “RFNs”; Tsai, et al., 2014), which requires dimerization mediated by offset sgRNA pairs, reduces off- target mutagenesis even more than paired nickases and truncated sgRNAs.
By contrast, off-target activity appears to be less of an issue for TALEN. Typically, TALENs are built with 18 repeats of 34 amino acids. The repeat vary at amino acids 12 and 13, the “Repeat Variable Diresidue”, or RVD. A DNA binding code mediated by the RVD (Figure 3) provides DNA binding specificity. A TALEN pair must bind on opposite sides of the target site, separated by a “spacer” ranging from 14-20 nucleotides (Figure 4). This offset design is necessary because FokI requires dimerization for activity. Therefore, such an extremely long (approximately 36 bp) DNA binding site is expected to be found rarely, if ever, in genomes. There is some degeneracy in the RVD-DNA binding code (Bogdanove and Voytas, 2011), but little evidence of mismatch tolerance or off-target activity has been demonstrated for TALEN. For example, in a recent study, an IPS cell line was edited with a highly active TALEN, and no mutagenic activity was detected at other genome sites homologous to the target site (Park, et al.,2014).

Figure 3. DNA binding code for TALENs. From Bogdanove & Voytas (2011).

Figure 4. Typical TALEN design.
2. Target site selection
As mentioned above, CRISPR sgRNA targets must immediately precede an N-G-G- site. It is usually not difficult to locate GG sites for knockouts, but this constraint sometimes causes problems for other applications. In addition, double nickase and RFN strategies have additional target design constraints. For both, two guide RNAs must be oriented such that the PAM sites are distal from to each other (“tail- to-tail” orientation; Mali, et al. 2013; Ran, et al., 2013; Shen, et al. 2014; Tsai, et al., 2014). Further, the spacing between the sgRNA tails is a critical factor influencing CRISPR design. Paired nickases work best with offset distances between -8 and 30 bp. Optimal spacing between 0 and 20 bp. RFN spacing is even more constrained. 13-18 bp spacing is required, with optimal spacing occurring at 16-17 nucleotides. Combined with the requirement for the PAM sites, the paired CRISPR strategies are sometimes difficult to implement for some applications.
On the other hand, while TALEN design requires offset binding proteins with defined spacing, no other design constraints have yet been described. So in principle, a TALEN pair can be targeted to any site in a genome, which should provide more freedom and flexibility in target site selection than for CRISPR.
3. Efficiency
CRISPR is popular partly because it is capable of modifying chromosomal targets at high frequencies. Rates of indel formation of more than 70% have been reported. The truncated sgRNAs, paired nickases, and RFNs usually have, as expected, lower efficiencies of indel formation.
TALEN is also able to modify chromosomes with high efficiency. In the example cited above (Park, et al.,2014), one TALEN pair had a measured indel formation of 33%. Such efficiency of indel generation is comparable to all CRISPR versions. TALEN, however, is sensitive to cytosine methylation, especially at CpG dinucleotides, which is a common and well-known mechanism for DNA silencing. CpG methylation occurs most often in promoter regions, but it can also occur in protein-coding DNA. Some TALEN pairs provide little to no mutagenesis activity, and this sensitivity to methylation is the likely reason.
4. Ease of design and construction
CRISPR is simple to design and use. For each target site, all that is needed is to program a 20 nucleotide genomic target site into the overall sgRNA. Plasmid construction is straightforward and simple. For editing experiments, the sgRNA is co-expressed with the re-usable Cas9 nuclease. sgRNA design and construction is identical for the paired nickase and RFN approaches, although in these latter cases, the sgRNAs must be chosen and designed to function as a coordinated pair.
1st generation CRISPR has a reputation that target designs are almost always successful. This is not true of the truncated sgRNAs, paired nickases, or RFNs. Some designs are successful, while others are not, even when designed in accordance with published data. Also, CRISPR is not methylation- sensitive.
Even though TALEN construction involves re-engineering a new protein for each target, TALEN design has been streamlined by the availability of modules of repeat combinations that reduce the amount of cloning. Also, as mentioned, TALEN is sensitive to cytosine methylation. One solution is to use the N* RVD for 5-methyl cytosine (Figure 3), but one must know a priori that the target site is methylated. Another workaround for this issue might is to avoid CpG sites. This would add another constraint, of course, but if successful would make TALEN even more attractive as a genome editing tool.
Which should I choose, TALEN or CRISPR?
CRISPR is a newer technology, but TALEN still provides a valuable option, thanks to its relatively unconstrained target site requirements and high degree of specificity. When deciding whether to use TALEN or CRISPR, it is critical to understand the differences between the two systems (Table 1). You should also consider the type of application you are attempting. For example, if you are in a hurry to knock a gene out as part of a quick pilot study, then first-generation CRISPR is a great choice. Need less off-target mutagenesis with relatively unconstrained design? Try TALEN.

Table 1. Comparisons between TALEN and CRISPR.
At GeneCopoeia, we provide products and services for both CRISPR and TALEN. Our genome editing begins with expert design of TALEN and CRISPR vectors in both non-viral and lentiviral formats. In addition, we offer donor plasmid design for HR mediated applications. Plasmid design is followed by construction and delivery of ready-to-use, sequence verified plasmid DNA. Further, we provide more sophisticated genome editing services, such as target site validation, and use of genome editing technology for stable cell line and transgenic mouse line generation. We also offer scientific consulting services, with which you can take advantage of our extensive knowledge and experience in the field. We will help you choose the option most appropriate for your experiments, by informing you of the differences described in this document as well as others, and work with you to devise sound genome editing strategies.
To learn more, visit our genome editing website:
https://www.genecopoeia.com/product/genome-editing/.
References
Bogdanove & Voytas (2011). TAL Effectors: Customizable proteins for DNA targeting. Science 333, 1843.
Fu, et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology 31, 822.
Fu, et al. (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnology 32, 279.
Li, et al. (2013). Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 31, 681.
Jinek, et al. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816.
Kuscu, et al. (2014). Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nature Biotechnology.
Mali, et al. (2013). Cas9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnology 31, 833.
Park, et al. (2014). Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Scie. U.S.A.
Ran, et al. (2013). Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 154, 1380.
Shen, et al. (2014). Efficient genome modification by CCRISPR-Cas9 nickase with minimal off-target effects. Nature Methods 11, 399.
Tsai, et al. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing van der Oost, et al. (2013). New Tool for Genome Surgery. Science 339, 768.
Wang, et al. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80.
Yang, et al. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas- mediated genome engineering. Cell 154, 1370.
Copyright ©2014
GeneCopoeia, Inc.
www.genecopoeia.com
TNGE4-062014 |
by admin | Aug 27, 2014 | Resources
|
|
Ed Davis, Ph.D. |
Genome Editing-the ability to make specific changes at targeted genomic sites-is of fundamental importance in biology and medicine (for reviews, see Bogdanove & Voytas, 2011; van der Oost, et al.,2013). Two genome editing technologies have emerged recently that exploit bacterial systems for plant pathogenesis or adaptive immunity: TALEN (Transcription Activator-Like Effector Nucleases) and CRISPR (Clustered, Regularly Interspaced, Short Palindromic Repeats), respectively. Both TALEN and CRISPR use endonucleases that initiate double-strand breaks (DSBs) at virtually any genomic target sequence, and can be used for many applications, including gene knock out, transgene knock in, gene tagging, and correction of genetic defects. However, researchers are often unaware of some of the work required to identify their desired modification in their cell lines. In this Technical Note, we discuss what you need to do for genome editing in mammalian cell culture after you have obtained your reagents from GeneCopoeia, the so-called “Downstream work”.
Upon receipt of plasmids
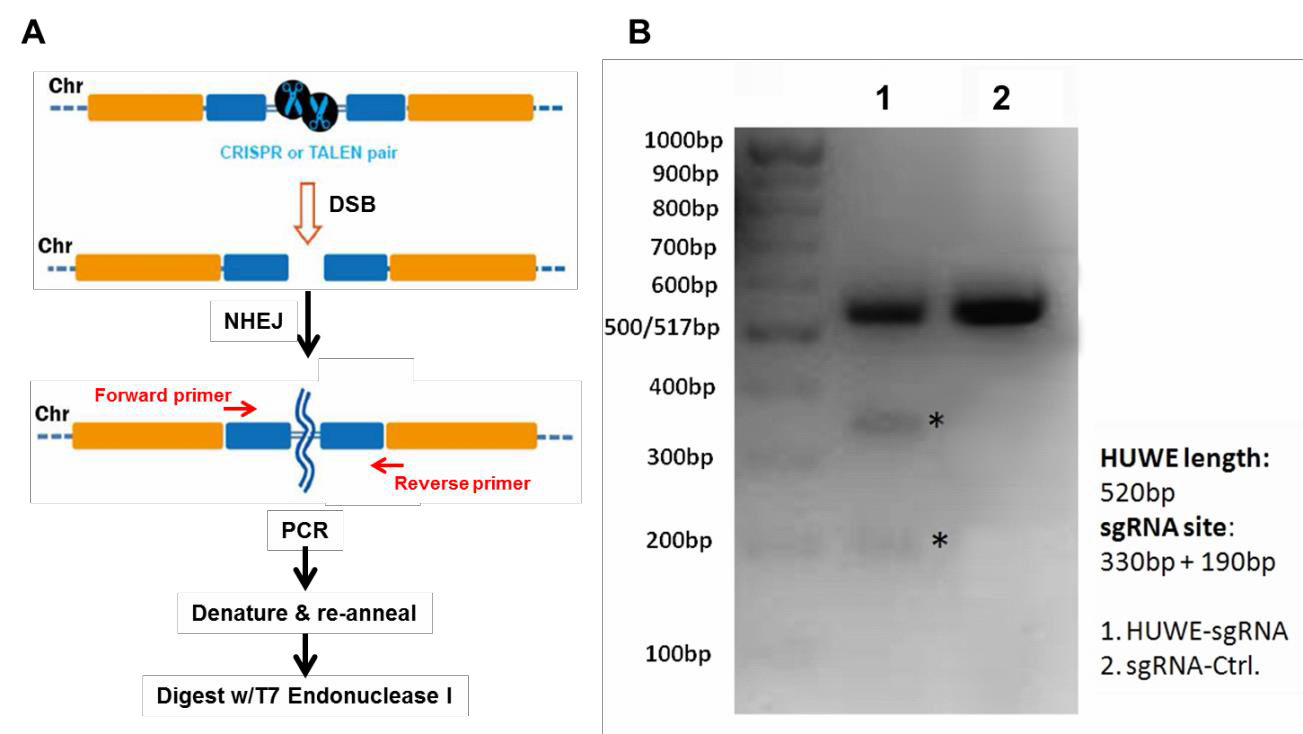
We recommend a specific workflow for how to use your GeneCopoeia genome editing plasmids (Figure 1). First, transform E. coli and make a frozen stock, so you’ll have an easily-renewable supply of DNA.
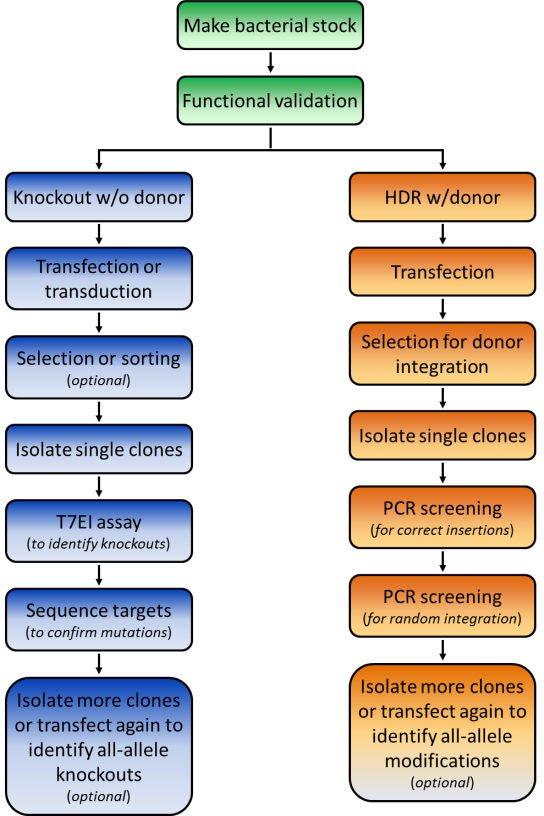
Figure 1. Pathways for repair of DSBs induced by genome editing tools. Left: Non-homologous end joining. Right: HR in the presence of a donor template.
|
|
Our plasmids are Ampicillin-resistant and can transform E. coli using standard protocols. Next, prepare a large amount of transfection-ready DNA using a commercial kit.
Functional validation
Before starting your experiments we recommend functional validation. This is optional, but will give you a good idea of how well your TALENs or CRISPRs function. We also offer functional validation in human HEK293T and mouse Neuro2a cells. You can also carry out the validation assays yourself in your chosen cell line.
For functional validation, we recommend using the mismatch cleavage assay (Qiu, et al., 2004). with T7 Endonuclease I. This assay looks for the presence of insertion or deletion (“indel”) mutations resulting from nonhomologous end joining (NHEJ) of DSBs at the target site.
The basic steps of this assay are outlined in Figure 2A. Briefly, cells are transiently transfected using standard methods, or transduced if using lentivral particles. Next, cells are harvested and used to prepare genomic DNA,followed by PCR using primers flanking the DSB site.
|
Finally, the PCR products are denatured, re-annealed, and digested with T7 Endonuclease I, which cleaves double stranded DNA containing mismatches. If cleavage was successful, then an agarose gel of the reaction products will show the presence of three bands: The full-length, undigested homoduplex band, and two smaller bands that are mismatch cleavage products (Figure 2B).
Figure 2. Mismatch cleavage assay. A. General scheme beginning with DSB formation, through digestion of mismatched PCR products with T7 Endonuclease I. B. Agarose gel of T7 endonuclease I digestion products. Lane 1 shows undigested as well as digested products, indicating successful cleavage. Lane 2 shows the reaction from a transfection using a scrambled negative control sgRNA.
Transfection or Transduction
Our non-viral plasmids for TALEN and CRISPR can be transfected by standard methodologies GeneCopoeia offers our EndofectinTM brand of transfection reagents. Alternatively, you can use whichever method works best for your cells. Our lentiviral CRISPR plasmids are compatible with 3rd- generation lentiviral packaging reagents. We also offer our own brand of LentifectTM lentiviral packaging reagents, as well as lentiviral packaging services.
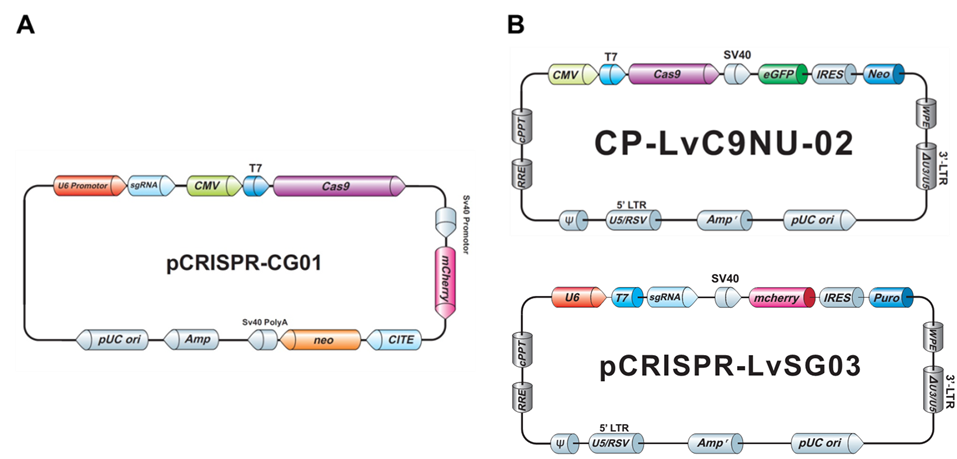
Both our non-viral as well as the lentiviral plasmids typically carry drug selection markers and fluorescent reporter genes, to aid you in your transfection and transduction procedures (Figure 3).
Clonal isolation
In genetics, it is essential to purify mutants away from a population-a practice known as clonal isolation. Clonal isolation minimizes the potential effects of unwanted modifications resulting from cell division or off-targeting, which could confound your results. When you are ready to begin screening, you’ll either need to plate cells such that they will form individual colonies, which you can then pick off the dish, or do serial dilutions in 96-well plates.
Figure 3. Examples of GeneCopoeia genome editing plasmids. A. Non-viral all-in-one plasmid containing CRISPR Cas9 nuclease, one CRISPR sgRNA, the neo gene for G418 selection, and mCherry as a fluorescent reporter. B. Lentiviral plasmids. Top, CRISPR Cas9 nuclease vector with the neo gene and eGFP. Bottom, CRISPR sgRNA plasmid with the puro gene for puromycin selection and mCherry.
Screening
The type of screening required depends on the type of genomic modification you are making. Three common situations are discussed:
1. Knockout by NHEJ
The fastest way to screen for NHEJ-mediated knockouts is by using the mismatch cleavage assay, as described above for functional validation. Once modified clones are identified, you’ll need to perform Sanger sequencing on the PCR products, to determine the exact nature of the mutation.
2. HR applications using a donor plasmid
The frequency of homologous recombination mediated by TALEN or CRISPR is typically much less than that of NHEJ, so it might seem you’d need to screen even more colonies to identify a modification with a donor plasmid. However, a donor plasmid provides drug selection, and, if present, a fluorescent reporter. GeneCopoeia donor plasmids carry drug selection and fluorescent reporter genes (Figure 4). After transfection, cells are plated in the presence of a drug, like puromycin, at low density to allow formation of single colonies. Alternatively, serial dilutions can be done in 96 well plates. Colonies that grow in the presence of the drug are candidates for stable cell lines carrying the modification.
Once drug resistant and/or fluorescent reporter-expressing clones are identified, the screening procedure differs dramatically from that used for NHEJ. For knockouts, the selection/reporter cassette is typically integrated in the protein-coding region, sometimes with a small deletion at the site of cleavage by TALEN or CRISPR. In other cases, a defined chromosomal region of DNA might be deleted and replaced with the cassette, or a gene might carry an in-frame fusion tag.
For screening, PCR with primers spanning the junction between the unmodified chromosome sequence and the sequence being inserted should be used. On both the left and right side of the insertion, one primer is designed for the homology arm, for synthesis in the direction of the marker, while the opposite primer targets the insertion for synthesis toward the homology arm. If the integration was successful, you will then be able to visualize bands of the expected sizes on an agarose gel.
The fastest way to screen for NHEJ-mediated knockouts is by using the mismatch cleavage assay, as described above for functional validation. Once modified clones are identified, you’ll need to perform Sanger sequencing on the PCR products, to determine the exact nature of the mutation.
The frequency of homologous recombination mediated by TALEN or CRISPR is typically much less than that of NHEJ, so it might seem you’d need to screen even more colonies to identify a modification with a donor plasmid. However, a donor plasmid provides drug selection, and, if present, a fluorescent reporter. GeneCopoeia donor plasmids carry drug selection and fluorescent reporter genes (Figure 4). After transfection, cells are plated in the presence of a drug, like puromycin, at low density to allow formation of single colonies. Alternatively, serial dilutions can be done in 96 well plates. Colonies that grow in the presence of the drug are candidates for stable cell lines carrying the modification.
Once drug resistant and/or fluorescent reporter-expressing clones are identified, the screening procedure differs dramatically from that used for NHEJ. For knockouts, the selection/reporter cassette is typically integrated in the protein-coding region, sometimes with a small deletion at the site of cleavage by TALEN or CRISPR. In other cases, a defined chromosomal region of DNA might be deleted and replaced with the cassette, or a gene might carry an in-frame fusion tag.
For screening, PCR with primers spanning the junction between the unmodified chromosome sequence and the sequence being inserted should be used. On both the left and right side of the insertion, one primer is designed for the homology arm, for synthesis in the direction of the marker, while the opposite primer targets the insertion for synthesis toward the homology arm. If the integration was successful, you will then be able to visualize bands of the expected sizes on an agarose gel.

Figure 4. Example of a GeneCopoeia donor plasmid for HR applications.
Using drug selection can lead to random integration of the donor plasmid, which must be ruled out. GeneCopoeia donor vectors carry the herpes simplex virus (HSV) thymidine kinase (TK) gene outside the region of recombination (Figure 4). If ganciclovir is included in the selection medium, then cells containing random integration of the donor plasmid will usually die, leaving a population enriched for those containing correct donor integration. Less frequently, random integration can exclude the TK gene, so PCR and/or Southern blotting should still be employed to rule out such events.
Finally, a gene modified by HR needs to be sequenced, so that both the chromosome structure surrounding the modified site is verified, and to detect the presence of an introduced mutation, if applicable.
At GeneCopoeia, our Genome Editing team has a wealth of expertise and experience in the design, construction, and use of TALEN and CRISPR reagents in mammalian systems. We start at TALEN and CRISPR design, delivering you sequence-verified plasmid DNA. We also offer functional validation services for your TALEN and CRISPR constructs. Further, we can construct stable cell lines or transgenic mice containing your TALEN- or CRISPR-mediated modification of interest. We also offer scientific consulting services to help you plan your genome editing projects. For more information, visit our website: https://www.genecopoeia.com. You can also call us at 1-301-762-0888, or email inquiry@genecopoeia.com
Things to look out for
Before devising your transfection and screening strategy, you should consider several factors that will affect the number of colonies you need to screen and, if necessary, the number of rounds of transfections needed to be carried out:
- Transfection efficiency. The percentage of cells that receive the TALEN or CRISPR reagents is critical for determining the number of colonies that need to be screened. If the transfection efficiency of your cell line is 40%, then you should expect to screen twice as many colonies as you would in a cell line with 80% transfection efficiency. If your cell line’s transfection efficiency is low, you can enrich for transfected cells by sorting using a fluorescent marker on one of the plasmids. You can also use drug selection for a short time period. We recommend estimating the transfection efficiency of your cell line using a plasmid that constitutively expresses a fluorescent reporter gene such as GFP.
- Copy number. For complete knockouts or mutagenesis, you might need to modify all alleles of the gene. Many people assume that they need to modify two alleles in a diploid cell line. However, many popular immortalized cell lines, such as HeLa, are aneuploid or polyploid, and so could contain more than 2 copies of your gene. The efficiency of modifying a single allele is higher than modifying multiple alleles in one cell. Therefore, if you need more than two alleles modified, you’ll need to screen more colonies than you would for a double allele modification. You might need to perform multiple rounds of transfection.
- Cutting efficiency. The efficiencies of indel formation by TALEN and CRISPR typically range from approximately 5% to about 70%. As with transfection efficiency, you should expect to screen twice as many colonies when the cutting efficiency is 20% as you would when the cutting efficiency is 40%.
The combination of factors will influence the number of colonies you’ll need to screen in order to find one with your modification. Simplistically, if you want a double-allele knockout in a diploid cell line using a CRISPR with 50% cutting efficiency and your transfection efficiency is 60%, then the percentage of colonies with a single allele modified would be 0.5 x 0.6 = 30%. For double allele modification, the percentage of positive colonies would be 15%. In this scenario, you’d need to screen at least 20 colonies to find one containing a double allele modification. If the transfection and cutting efficiencies are even lower, and your cell line contains more than two copies of your target, you will need to screen many more colonies in order to find one with a complete knockout.
References
Bogdanove & Voytas (2011). TAL Effectors: Customizable proteins for DNA targeting. Science 333, 1843.
Qiu, et al. (2004). Mutation detection using Surveyor™ nuclease. Biotechniques 36, 702.
van der Oost, et al. (2013). New Tool for Genome Surgery. Science 339, 768.
Copyright ©2014
GeneCopoeia, Inc.
www.genecopoeia.com
TNGE3-070914 |
by admin | Aug 27, 2014 | Resources
 |
|
Ed Davis, Ph.D. |
Abstract
Genome Editing-the ability to make specific changes at targeted genomic sites in complex organisms-is of fundamental importance in biology and medicine (Bogdanove & Voytas, 2011; van der Oost, et al.,2013). Recently, the CRISPR (Clustered, Regularly Interspaced, Short Palindromic Repeats)-Cas (CRISPR- associated) system has become popular for applications such as gene knockouts, making precise, defined base changes, and for transgenesis, to name a few. The ease of design, high efficiency, and relatively low cost of CRISPR-Cas offers promise for use of this tool for correcting mutations that cause genetic diseases, and to replace older methods that cause undesired consequences of random transgene integration. However, CRISPR-Cas itself has some propensity for causing off-target mutagenesis. Despite recent improvements in the technology, some researchers believe that CRISPR-Cas has a relatively low degree of specificity. In this Technical Note, we discuss the mechanism of CRISPR-Cas in its application for genome editing and how it affects specificity, reports in the literature discussing CRISPR’s potential for off-target mutagenesis, approaches for increasing the target specificity of CRISPR- Cas, and GeneCopoeia solutions for improving CRISPR-Cas specificity.
CRISPR-Cas target recognition
The most widely-used version of CRISPR-Cas in genome editing applications employs two components:
1) The Cas9 nuclease from Streptococcus pyogenes; and 2) A single guide RNA (sgRNA). The sgRNA is a chimera of two RNA molecules of the CRISPR target recognition system, as first described by Jinek, et al. (2012): The CRISPR RNA (crRNA) and the transactivating CRISPR RNA (tracrRNA; Figure 1). An sgRNA contains a 20 nucleotide variable region that provides target site specificity. In addition, the S. pyogenes Cas9 requires a 3 nt N-G-G sequence, known as a “PAM” (Protospacer Adjacent Motif), immediately following the specificity sequence, for DNA binding. The PAM must be on the same strand as the guide RNA sequence, but not part of it.

Figure 1. S. pyogenes Cas9 nuclease requires the presence of two RNAs, the target-specific crRNA, and the structural tracrRNA, for binding and cleavage. The target site is highlighted in yellow. The PAM site is highlighted in gray. Arrows indicate potential nuclease cutting sites. The red arrow is the most frequent cutting position. These RNAs can be fused into one sgRNA for genome editing. From Jinek, et al. (2012).
When the sgRNA forms a complex with Cas9, the complex locates the target and PAM sequence, unwinds the DNA duplex, and the 20nt guide RNA anneals to the complementary sequence on the opposite strand. This enables the Cas9 nuclease to create a double-strand break (DSB; Figure 1). DSBs must be repaired immediately or the cell will die. The cell’s preferred pathway of DSB repair is the relatively error-free mechanism of homologous recombination (HR), but this is almost never available in mitotically dividing cells because it must occur during anaphase, after chromosomes have duplicated and sister chromatids have paired. HR also usually doesn’t occur in non-dividing cells. Instead, in the absence of homology, DSB repair occurs via non-homologous end joining (NHEJ). NHEJ is extremely error-prone, leading to the formation of small insertions or deletions (indels) as the broken chromosome ends are pieced back together. This proclivity for indel generation has been exploited by researchers using CRISPR-Cas as a simple, efficient method for knocking genes out.

Figure 1. Pathways for repair of DSBs induced by genome editing tools. Left: Non-homologous end joining. Right: Homologous recombination in the presence of a donor template.
Specificity of sgRNA binding
A given CRISPR-Cas9 target sequence is 20 nt long, so, if binding of the RNA to the chromosomal target were based strictly on sequence, these targets should be unique in eukaryotic genomes. In combination with the, PAM, it should be extremely unlikely that CRISPR-Cas would make another cut.
But there’s more to CRISPR specificity than just sequence composition. Yanfang Fu and colleagues reported that sgRNAs can guide the Cas9 nuclease to three different targets in a chromosomally- integrated GFP reporter gene, despite the presence of one or more mismatches (Fu, et al., 2013).
Dispersed as well as adjacent multiple mismatches between the guide RNAs and the reporter target could be tolerated as well. Mismatches in the 10 nucleotides of the guide RNA most proximal to the PAM site were usually-though not always-tolerated to a greater degree than those in the PAM-distal 10 nucleotides, and the distribution tolerance of mismatches varied depending on the target.
In addition, Fu, et al., generated guide RNAs for three endogenous human genes and predicted, based on the reporter assay results, additional off-target sites in the genome for each. Nearly all off-target sites contained multiple mismatches with each target RNA. Using the T7 Endonuclease I assay to detect the presence of indels, Fu, et al. found that these predicted off-target sites frequently sustained indels, often as much or more than the intended target sites. Up to 5 mismatches between the guide RNA and the chromosome could still allow detectable indel formation. The degree of mismatch tolerance varied depending on the guide RNA sequence, the chromosomal targets analyzed, and the cell lines used.
Recent analysis of CRSIPR-Cas off-target mutagenesis
Early tests of CRISPR-Cas specificity such as those by Fu, et al. (2013) cast doubt on the viability of using this technology for applications requiring high specificity, such as gene therapy. Newer studies, though, have reported better specificity for CRISPR-Cas. For example, Eric Lander’s lab at MIT used pools of lentiviral based sgRNAs for large-scale mutagenesis screens in human cells. Screens for resistance to DNA damaging agents demonstrated that mutagenesis of genes in the mismatch repair pathway occurred at much greater frequencies by correctly-targeted sgRNAs than by incorrectly-targeted guides (Wang, et al., 2014). In addition, an sgRNA that was used to successfully restore wild-type function to the Crygc gene and cure cataracts in mice had no effect in 10 of 12 transgenic animals at 10 predicted off-target sites, and only 1 site was mutated in each of the remaining 2 mice (Wu, et al., 2013). These and other studies (Li, et al., 2013; Yang, et al., 2013), combined with improved software tools for predicting sgRNAs with high potential specificity (Ran, et al., 2013a; Xiao, et al., 2014), suggest that lingering worries that CRISPR-Cas9 has a high propensity for off-target mutagenesis might be overstated.
GeneCopoeia solution to CRISPR-Cas off-target mutagenesis
GeneCopoeia uses sophisticated bioinformatics tools to predict potential genomic sites for Cas9 sgRNA off-target binding. Whether the application is knockout, knockin, introduction of base changes, etc., we design CRISPR sgRNAs that are most likely to function properly, while at the same time discarding those guides with a high likelihood for causing off-target mutagenesis.
Cas9 variants for increased specificity
In addition to improved guide RNA design, mutant forms of Cas9 have emerged to further decrease CRISPR-mediated off-target mutagenesis. Wild-type Cas9 has two nuclease domains, each of which cut one DNA strand. Mutations in either domain alone convert Cas9 into a “nickase”, which makes only a single-strand break. While the nickase mutants do not change sgRNA binding specificity and can still be delivered to off-target sites, single nicks have much lower potential for causing mutagenic NHEJ than DSBs. However, when two guide RNAs are designed such that each binds the opposite strand of the chromosome, the result is a staggered-cut DSB that can be repaired by NHEJ or HR (Figure 2). Two groups-one led by George Church at Harvard and the other by Feng Zhang at MIT, showed that this “paired nickase” strategy efficiently led to DNA repair by both NHEJ and HR (Ran, et al., 2013b; Mali, et al., 2013). Zhang’s group further demonstrated that the paired nickase strategy dramatically reducedthe frequency of off-target mutagenesis from 50-fold to more than 1,500 fold for sgRNAs targeted to three distinct genes.

Figure 2. General scheme of Cas9 double-nickase strategy. From Ran, et al. (2013b).
Double nickase design is not easy, though. Guide RNAs oriented with the PAM sites most distal from one another (a “tail-to-tail” orientation; Shen, et al., 2014) lead to indel formation at much higher frequencies than those oriented with the PAM sites proximal to one another (“head-to-head”; Ran, et al., 2013b; Mali, et al., 2013). In addition, the spacing between the ends of each guide sequence (the offset distance) is important. The most efficient nickase pairs generally occur with offset distances between 0 and 20 base pairs. So while the double nickase approach works well to efficiently create DSBs with few consequences resulting from off-target cleavage, the design constraints limit the potential range of suitable sites for some applications.
Further, Keith Joung’s lab published two papers describing two new potential Cas9 alternative strategies that also might elevate the level of CRISPR-Cas specificity. One of the papers showed that sgRNA sequences with as few as 17 nucleotides were usually as effective at creating indels as the standard length of 20 nucleotides (Fu, et al., 2014). Off-target mutagenesis caused by these truncated sgRNAs targeted to four genes was reduced by up to 5,000 fold. In the other paper (Tsai, et al., 2014) Joung’s lab fused a Cas9 mutant with no nuclease activity (Cas9n) to the nuclease domain of FokI. FokI requires dimerization to cut, so, in a fashion similar to the double nickase strategy, a Cas9n-FokI pair was shown to dramatically reduce off-targeting. However, it remains to be seen whether the approaches of the truncated sgRNAs, which don’t always function at high efficiency, or the Cas9-FokI fusion, which has spatial design constraints, will be effective in general.
GeneCopoeia solutions for increased Cas9 specificity
GeneCopoeia scientists are experts at Cas9 D10A double nickase design strategy. We diligently predict the paired guide RNAs that have the highest probability of efficient cutting, based on standards reported in the literature, as well as our own proprietary requirements. Our double nickases are designed to function either as a pair with the Cas9 D10A nickase (which we provide) or singly using the wild type Cas9 nuclease, offering customers great flexibility. In addition, we also design truncated 17-, 18- and 19- mer sgRNAs for those customers who request it. We can also design sgRNAs pairs intended for use in the Cas9-FokI fusion approach.
Conclusions
Taken together, it’s clear that improvements in sgRNA design and strategy, combined with alternative versions of the Cas9 nuclease, make CRISPR-Cas9 a viable approach for many applications, including gene therapy, with much less concern about off-target mutagenesis than initially thought.
At GeneCopoeia, we offer a full suite of CRISPR-Cas9 genome editing tools, including both the wild-type and D10A nickase versions of Cas9. Our service includes sgRNA design for single and double nickase targets. We also carry sgRNA and Cas9 vectors in both lentiviral and non-viral formats. Finally, our services don’t stop at plasmid design and construction. We also generate stable cell lines and mouse lines carrying CRISPR-Cas9-mediated modifications, to meet all your genome editing needs.
References
Bogdanove & Voytas (2011). TAL Effectors: Customizable proteins for DNA targeting. Science 333, 1843.
Fu, et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology 31, 822.
Fu, et al. (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnology 32, 279.
Jinek, et al. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816.
Li, et al. (2013). Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 31, 681.
Mali, et al. (2013). Cas9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnology 31, 833.
Ran, et al. (2013a). Genome engineering using the CRISPR-Cas9 system. Nat. Protocols 8, 2281.
Ran, et al. (2013b). Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 154, 1380.
Shalem, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84.
Shen, et al. (2014). Efficient genome modification by CRISCRISCRISCRISPR-Cas9 nickase with minimal off target effects. Nature Methods 11, 399.
Tsai, et al. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing.
Nature Biotechnology Apr 25. doi: 10.1038/nbt.2908. [Epub ahead of print].
Wang, et al. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80.
Wu, et al. (2013). Correction of a Genetic Disease in Mouse via Use of CRISPR-Cas9. Cell Stem Cell 13,
659.
Xiao, et al. (2014). CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30, 1180.
Yang, et al. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas- mediated genome engineering. Cell 154, 1370.
Copyright ©2014
GeneCopoeia, Inc.
www.genecopoeia.com
TNGE2-050814 |
by admin | Aug 26, 2014 | Resources
|
Ed Davis, Ph.D. |
Abstract
Genome Editing-the ability to make specific changes at targeted genomic sites-is fundamentally important to researchers in biology and medicine (Bogdanove & Voytas, 2011; van der Oost, et al., 2013). Two genome editing technologies have emerged recently that exploit bacterial systems for plant pathogenesis or adaptive immunity: TALEN (Transcription Activator-Like Effector Nucleases) and CRISPR (Clustered, Regularly Interspaced, Short Palindromic Repeats), respectively. Both TALEN and CRISPR use endonucleases that initiate double-strand breaks (DSBs) at virtually any genomic target sequence, and are used for many applications, including gene knockout, transgene knock-in, gene tagging, and correction of genetic defects. In this Review, we discuss and compare genome editing technologies, applications for genome editing using published case studies, and how GeneCopoeia genome editing technologies and services can accelerate your research.
Introduction
What is Genome Editing?
In the strictest sense, genome editing means making stable, permanent, and heritable changes to the genetic code, to accomplish many potential goals. The process begins with stimulation of a DSB at the target site. DSBs are lethal if left unrepaired, so eukaryotic cells have several mechanisms in response (Figure 1). The first is homologous recombination (HR), whereby cells use a homologous copy of the broken chromosome as a repair template. HR is a relatively error-free process. The template is normally the sister chromatid during G2 in mitosis, but can also come from a DNA fragment introduced exogenously, which can mediate a “knock in” of desired DNA sequences to the target site.
|
|
Figure 1. Pathways for repair of DSBs induced by genome editing tools. Left: Non-homologous end joining. Right: HR in the presence of a donor template.
The second major mechanism for DSB repair is nonhomologous end joining (NHEJ), which occurs when no homologous template is available. NHEJ is simply the re-connection of the broken chromosome ends. However, NHEJ is error-prone, and frequently leads to small insertions or deletions (“indels”) at the break site. Indels can disrupt a gene by causing frame shifts, and, therefore, gene knockouts.
|
Genome Editing tools can also be used for non-permanent changes in gene expression, by adapting them as fusions to either transcriptional activators or repressors. While this is a popular application of these tools, it will not be covered in this review.
RNAi-mediated knockdown is the most common strategy for ablating gene function in higher eukaryotes. However, there are a few key differences between RNAi and genome editing. First, RNAi does not completely shut off the gene (Ketting, 2013). Rather, gene expression is down-regulated post- transcriptionally, without changing the genetic code (Mittal, 2004). Some functional RNA or protein remains and is translated. So, the RNAi strategy is a “knockdown”. Gene function is reduced, not eliminated. In genome editing, on the other hand, the genetic code is changed, and attenuation of gene expression is usually complete, leading to a “knockout”. The decision on whether to use RNAi or genome editing for attenuating gene expression depends on the goals of the experiment. (Table 1).
| Method |
Knock
down |
Knock
out |
genetic
code |
Change
expression
level |
Clone
isolation
required |
| RNAi
(shRNA, siRNA) |
√ |
|
|
√ |
|
| Genome editing
(TALEN, CRISPR) |
|
√ |
√ |
√ |
√ |
Table 1. Comparison between RNA interference and Genome editing methods for gene ablation.
Genome editing technologies
TALEN
In nature, TALs are DNA binding proteins from bacteria that infect plants (Bogdanove & Voytas, 2011). DNA binding is mediated by 34 bp amino acid repeats, which differ at amino acids 12 and 13. These “repeat variable diresidues”, or RVDs, facilitate TALEN DNA binding specificity. Each RVD binds one nucleotide, and a DNA binding code has been determined (Figure 2).
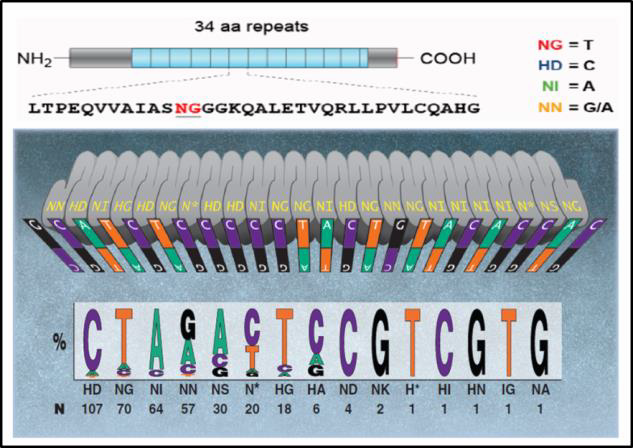
|
| Figure 2. DNA binding code for TALENs. Adapted from Bogdanove & Voytas (2011).
TALENs consist of two DNA binding proteins. Each is fused to one domain of the FokI restriction endonuclease, and recognizes 17-18 bp of target sequence. FokI requires dimerization for activity, so when two properly designed TALs-on opposite sides of the intended break site and on opposite strands,with approximately 18 nucleotides in between-come together, FokI cuts the DNA. The resulting DSB is repaired by either NHEJ or HR as described above.
|
CRISPR
CRISPR’s mechanism differs from TALEN, with the same end result. CRISPR’s natural function is to destroy phages that invade bacteria for adaptive immunity. For genome editing, the S. pyogenes Cas9 nuclease is inactive until it binds to a single guide RNA (sgRNA). An sgRNA contains a 20 nucleotide genomic sequence. The Cas9-sgRNA complex recognizes this sequence in the genome when it is immediately followed by a 5’ N-G-G 3’ “PAM” site. Upon recognition, the sgRNA hybridizes with the strand opposite the PAM, and Cas9 creates a DSB (Figure 3).
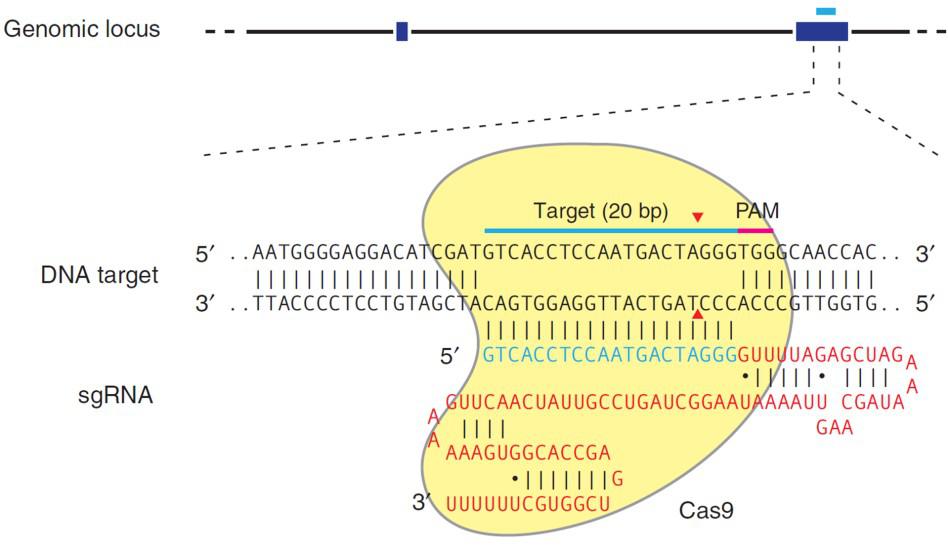
| Figure 3. Recruitment of Cas9 to a target locus by an sgRNA. Red sequences, sgRNA scaffolding. Blue, target sequence. The PAM is indicated by the red bar. The sgRNA target sequence binds to the strand opposite the PAM site. Cas9 cuts both sDNA strands between the third and fourth nucleotide 5’ of the PAM. Red arrows: Position of the DNA strand cuts. From Ran, et al. (2013). |
|
Applications for genome editing
Because genome editing begins with DSBs, many applications are possible. The most common is gene knockout. At the simplest level, either a TALEN pair, or the combination of the Cas9 nuclease with a single guide RNA, creates a DSB, leading to error-prone repair. Occasionally, small insertions occur, but NHEJ more commonly results in the formation of small deletions of varying lengths (Figure 3).
|
| Figure 4. TALEN activity leads to target site-specific insertions or deletions. From Wang, et al. (2013).
The frequencies of frameshifts vary widely, influenced by the particular TALEN pair or CRISPR sgRNA. Reports in the literature of efficiencies, as measured by the percentage of cell line clones carrying a mutation, range from 0% to more than 70%.
|
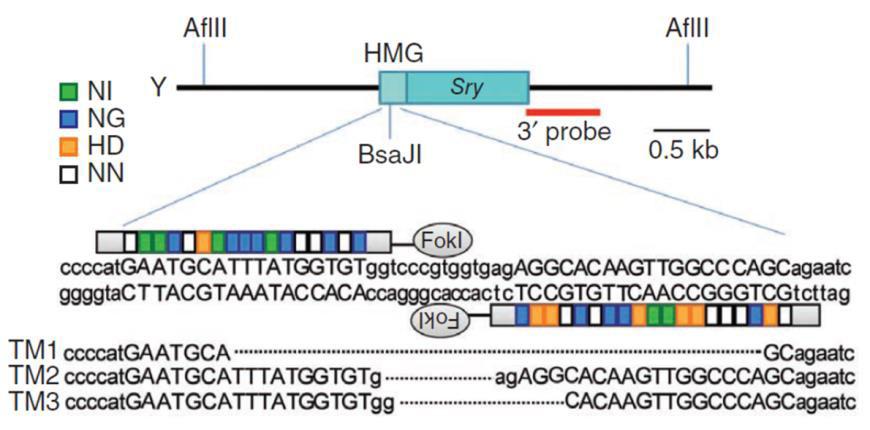
Knockouts can also be achieved with a homologous donor (Figure 5). In the simplest case, the sgRNA is designed to cut in the earliest protein-coding region in common to all splice variants. It can be the same target sequence as that used for donorless knockouts. The donor is constructed such that insertion of a selection/reporter cassette causes a deletion in the sgRNA binding site, to both knock the gene out and simultaneously prevent cleavage of the donor. The donor is co-introduced with the Cas9/sgRNAs. 2-3 days post-transfection, selection for the drug resistance gene in the donor (e.g. puromycin) is applied, single drug-resistant colonies are identified, and then screened for the correct insertions.
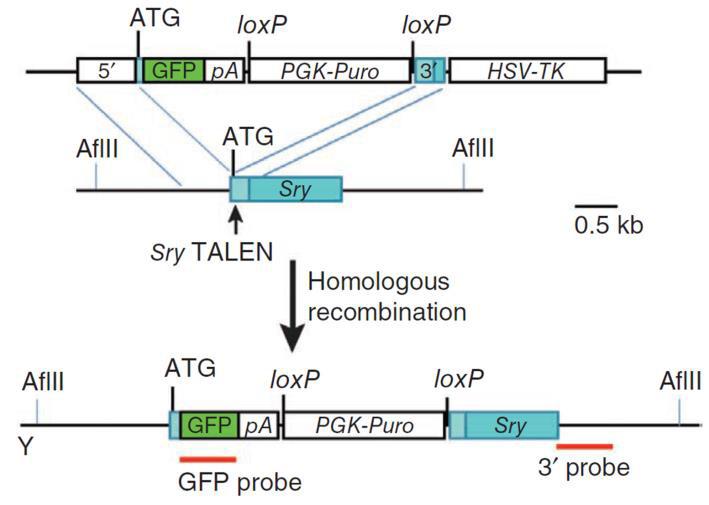
|
|
Figure 5. Knockout of the mouse Sry gene using a donor plasmid. Top. Donor plasmid consisting of a cassette with GFP and a puromycin resistance gene. The cassette is flanked by sequences homologous to the mouse Sry target region. The clone is designed such that the cassette is inserted between the initiator ATG and the rest of the gene. Bottom. Co-introduction of a TALEN with the donor plasmid causes recombination between the donor and the Sry locus, leading to replacement of the endogenous sequences with the sequence interrupted by the cassette and knockout of the gene. From Wang, et al. (2013)
|
GeneCopoeia carries all the reagents necessary for customers to knock their genes out, by either NHEJ- mediated indels, or with a donor clone. These include clones expressing TALEN pairs or sgRNAs targeted to a region if interest, Cas9 clones, and donor clones built for knockout as well as other applications. Our CRISPR clones come in non-viral and lentiviral format, for hard-to-transfect cell lines.
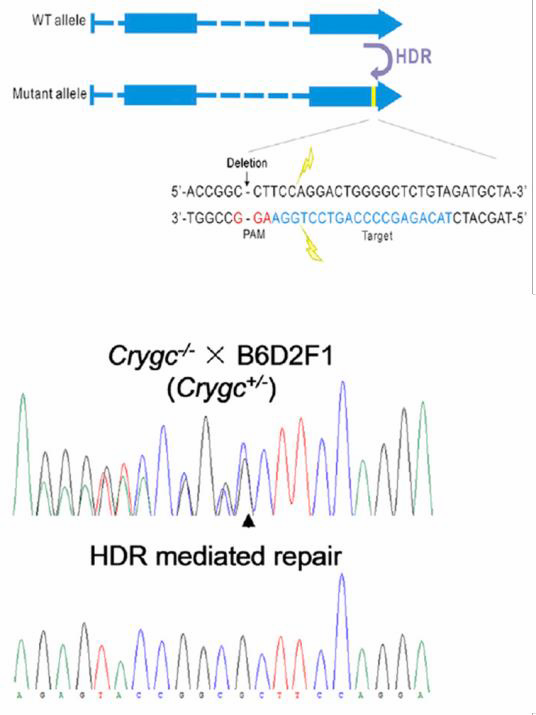
| Figure 6. Curing of heritable cataract disorder using CRISPR
and an ssODN in transgenic mice. From Wu, et al. (2013)
Other genome editing applications require a homologous donor. One is introduction of a single base change, which can be achieved using a donor plasmid or a single strand
oligonucleotide (ssODN). In the ssODN strategy, an oligonucleotide carries the base change and 40-90 nucleotides of homology flanking the target region. The method works with high efficiency. Jinsong Li’s lab used CRISPR with an ssODN to correct a single base mutation that causes cataracts in mice (Wu, et al., 2013; Figure 6).
|
|
Which should I use, TALEN or CRISPR?
Both TALEN and CRISPR are highly effective technologies for genome editing. Each, however, has its limitations, and these factors need to be taken into account when choosing one technology over the other. On one hand, TALEN is sensitive to cytosine methylation, and so a methyl C-specific RVD must be designed for recognition. CRISPR, though, is not methylation- sensitive. In addition, TALEN cutting efficiency tends to be lower than CRISPR’s. On the other hand, TALEN tends to be less prone to off-target mutagenesis than CRISPR, although recent improvements in the technology, such as the use of double nickases, truncated guide RNAs, and a Cas9-Fok I fusion, improve CRISPR target site specificity.
Conclusions
At GeneCopoeia, we provide a full suite of genome editing products and services. These begin with TALEN and CRISPR clone design and construction, but our expertise doesn’t stop there. We also offer functional validation of TALEN and CRISPR clones. In addition, we will construct both stable cell lines and transgenic mice carrying your modification of interest. From our long-standing expertise in mammalian ORF and promoter clones, we have built a database of more than 40,000 knockout targets for human and mouse, which can be conveniently purchased from our website. Our experts can also help you with demanding custom genome editing applications. Contact us today at
inquiry@genecopoeia.com!
For more information, please visit our Genome Editing page:
https://www.genecopoeia.com/product/genome-editing/
References
Bogdanove & Voytas (2011). Science 333, 1843.
Ran, et al. (2013). Nature Protocols 8, 2281.
van der Oost, et al. (2013). Science 339, 768.
Wang, et al. (2013). Nature Biotech. 31, 530.
Wang, et al. (2014). Science 343, 80.
Wu, et al. (2013). Cell Stem Cell 13, 659.
| Copyright ©2014
GeneCopoeia, Inc.
www.genecopoeia.com
RAGE1-080614 |
by admin | Aug 26, 2014 | Resources
|
|
Ed Davis, Ph.D. |
Recent advances in technologies for genome editing-the use of TALEN or CRISPR to make targeted, permanent changes to genes-have revolutionized molecular genetics. They have also presented users with a choice between these relatively new technologies and that of the more established method of RNA interference (RNAi)-mediated knockdown using short hairpin RNA (shRNA) or short interfering RNA (siRNA). In this Technical Note, we explore the differences between the two methods for ablating gene function, and situations where one technology is more appropriate than the other.
RNAi-mediated gene silencing
In higher eukaryotes, RNAi-mediated knockdown is the most common strategy for depleting cells of a gene product of interest. However, RNAi usually does not completely shut off the gene. Essentially, short (approximately 20-25 nucleotides) double stranded RNA molecules are either generated from hairpin-forming precursors (shRNAs) or introduced exogenously (siRNAs). After processing by Dicer, a single stranded RNA base-pairs with a target mRNA (Ketting, 2013). Depending on the organism, RNAi- mediated gene silencing is carried out by Argonaute proteins via either mRNA degradation or translation inhibition (Figure 1). The end result is post transcriptional down-regulation of gene expression, without changing the genetic code (Mittal, 2004). Some functional RNA or protein remains and is translated at lower levels. So, the RNAi strategy for reducing gene function is termed a “knockdown”. Gene function is reduced, but not eliminated.

Figure 1. General scheme of RNAi pathways. From Mittal (2004).
Genome editing for gene knockout
By contrast, genome editing changes the genetic code, typically causing a “knockout”, or complete elimination of gene function. The process begins with creation of a double-strand break (DSB) in the chromosome (Bogdanove & Voytas, 2011). Recently, two tools have been developed for generating DSBs with high efficiency: Transcription Activator-Like Effector Nucleases (TALENs), and Clustered, Regularly Interspaced Palindromic Repeat Associated (CRISPR-Cas) proteins (Bogdanove & Voytas, 2011; Jinek, et al., 2012; Shalem, et al., 2014; Wang, et al., 2014). Both these tools are adapted from bacterial systems that either cause plant pathogenesis (TALEN), or defend the genome from insertional mutagenesis (CRISPR-Cas). TALENs are chimeric proteins consisting of site-specific DNA binding proteins fused to the restriction endonuclease FokI. CRISPR-Cas uses a site-specific, 20 nucleotide single guide RNA (sgRNA) to bring the Cas9 nuclease to its target locus. For both TALEN and CRISPR-Cas, the nuclease cuts both DNA strands of the target. This break must be repaired or the cell will die, so eukaryotic cells respond by two major mechanisms (Figure 2). The first, non-homologous end joining (NHEJ), re-ligates the two free chromosome ends. However, NHEJ is error-prone, often resulting in small insertions or deletions that can disrupt, or knock out, the gene. Alternatively, cells can repair DSBs through homologous recombination (HR), which provides researchers more options for gene knockout. Defined deletions can be introduced, insertional mutations can be created, or single bases can be changed, to name a few.

Figure 2. Pathways for repair of DSBs induced by genome editing tools. Left: Non-homologous end joining. Right: Homologous recombination in the presence of a donor template.
Comparisons between RNAi and genome editing
So, which strategy is better: RNAi-mediated knockdown, or genome editing-mediated knockout? This depends on the experimental goals (Table 1). Indeed, some people confuse the two strategies, referring to genome editing as “knockdown”. This confusion is likely due to the fact that for many years before genome editing became feasible, the most practical strategy for ablating gene function in higher eukaryotes was using RNAi. Thus, researchers became acclimated to the term “knockdown”.
| Method |
Knock
down |
Knock
out |
genetic
code |
Change
expression
level |
Clone
isolation
required |
RNAi
(shRNA, siRNA) |
√ |
|
|
√ |
|
Genome editing
(TALEN, CRISPR) |
|
√ |
√ |
√ |
√ |
Table 1. Comparison between RNA interference and Genome editing methods for gene ablation.
Basically, RNAi-mediated knockdown is preferable to genome editing when changing the genetic code is undesirable. For example, you might want to reduce gene function temporarily, so you could transiently transfect siRNAs into cells. In a few generations, the siRNAs are lost, restoring normal gene function. Alternatively, you can stably integrate shRNAs into the genome and express them from an inducible promoter. Then you can turn the expression of the gene down and then back up repeatedly, at desired times, and/or in specific tissues. Moreover, shRNA-mediated knockdown does not require the isolation of single clones, unlike genome-editing mediated knockout, so there is less work involved. Finally, completely eliminating gene function might harm the cell, but a partial loss will not.
Alternatively, to make a true genetic null allele, genome editing is preferable (Wang, et al., 2013). Additionally, one might want to introduce a specific point mutation, or correct a pre-existing mutation back to wild type. Finally, you might want to add a fusion tag to your favorite gene and express it from its endogenous locus. These goals absolutely depend on genome editing methods.
GeneCopoeia offers advanced, full-service solutions for both RNA interference and genome editing, from design and construction of shRNAs and TALEN or CRISPR plasmids, all the way up to the generation of stable cell lines and transgenic mice. Please visit our website to learn more: www.genecopoeia.com.
References
Bogdanove & Voytas (2011). TAL Effectors: Customizable proteins for DNA targeting. Science 333, 1843.
Jinek, et al. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816.
Ketting (2011). The many faces of RNAi. Dev. Cell 20, 148.
Mittal (2004). Improving the efficiency of RNA interference in mammals. Nat. Rev. Genet. 5, 355.
Shalem, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84.
Wang, et al. (2013). TALEN-mediated editing of the mouse Y chromosome. Nature Biotech. 31, 530.
Wang, et al. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80.
Copyright ©2014 GeneCopoeia, Inc.
Email: inquiry@genecopoeia.com
Tel: +1 (866) 360-9531
TNGE1-021814 |